
ENHERTU 100 mg, poudre pour solution à diluer pour perfusion, boîte de 1 flacon de 5 ml de solution reconstituée
Dernière révision : 31/03/2025
Taux de TVA : 2.1%
Laboratoire exploitant : DAIICHI SANKYO FRANCE
Source :

Cancer du sein
Cancer du sein HER2-positif
Enhertu en monothérapie est indiqué dans le traitement des patients adultes
présentant un cancer du
sein HER2-positif non résécable ou métastatique ayant reçu préalablement au
moins une ligne de traitement anti-HER2.
Cancer du sein HER2-faible et HER2-ultrafaible
Enhertu en monothérapie est indiqué dans le traitement des patients adultes
présentant :
- un cancer du sein non résécable ou métastatique avec récepteurs hormonaux positif (RH+), HER2-faible ou HER2-ultrafaible ayant reçu au moins une hormonothérapie au stade métastatique et qui ne sont pas éligibles à une hormonothérapie en ligne de traitement suivante (voir rubriques Posologie et mode d'administration et Propriétés pharmacodynamiques) ;
- un cancer du sein HER2-faible non résécable ou métastatique ayant reçu préalablement une chimiothérapie pour la maladie métastatique ou ayant présenté une récidive de la maladie pendant la chimiothérapie adjuvante ou au cours des six mois suivant la fin de la chimiothérapie adjuvante (voir rubrique Posologie et mode d'administration).
Cancer bronchique non à petites cellules (CBNPC)
Enhertu en monothérapie est indiqué dans le traitement des patients adultes présentant un CBNPC de stade avancé avec mutation activatrice du gène HER2 (ERBB2) nécessitant un traitement systémique après une chimiothérapie à base de platine associée ou non à une immunothérapie.
Cancer de l'estomac
Enhertu en monothérapie est indiqué dans le traitement des patients adultes présentant un adénocarcinome gastrique ou de la jonction œsogastrique (JOG) HER2-positif de stade avancé ayant reçu préalablement une ligne de traitement comportant le trastuzumab.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Afin d'éviter des erreurs médicamenteuses, il est important de vérifier l'étiquette du flacon pour s'assurer que le médicament préparé et administré est Enhertu (trastuzumab déruxtécan) et non le trastuzumab ou le trastuzumab emtansine.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Pneumopathie interstitielle diffuse/pneumopathie inflammatoire
Des cas de pneumopathie interstitielle diffuse (PID)/pneumopathie inflammatoire ont été rapportés avec Enhertu (voir rubrique Effets indésirables). Des issues fatales ont été observées. Les patients doivent être informés qu'ils doivent signaler immédiatement toute toux, dyspnée, fièvre et/ou toute apparition ou aggravation de symptômes respiratoires. Les patients doivent être surveillés afin que les signes et symptômes de PID/pneumopathie inflammatoire puissent être détectés. Des investigations doivent être réalisées rapidement en cas de signes de PID/pneumopathie inflammatoire. Des examens d'imagerie, de préférence par tomodensitométrie (TDM), doivent être réalisés en cas de suspicion de PID/pneumopathie inflammatoire. La consultation auprès d'un pneumologue doit être envisagée. En cas de PID/pneumopathie inflammatoire asymptomatique (grade 1), une corticothérapie (par exemple ≥ 0,5 mg/kg par jour de prednisolone ou équivalent) doit être envisagée. Le traitement par Enhertu doit être interrompu jusqu'à la résolution au grade 0 et peut être repris conformément aux instructions figurant dans le tableau 2 (voir rubrique Posologie et mode d'administration). En cas de PID/pneumopathie inflammatoire symptomatique (grade ≥ 2), une corticothérapie (par exemple ≥ 1 mg/kg par jour de prednisolone ou équivalent) doit être instaurée rapidement et poursuivie pendant au moins 14 jours, avec ensuite une diminution progressive de la dose sur 4 semaines au moins. Le traitement par Enhertu doit être arrêté définitivement chez les patients chez lesquels une PID/pneumopathie inflammatoire symptomatique (grade ≥ 2) est diagnostiquée (voir rubrique Posologie et mode d'administration). Les patients ayant des antécédents de PID/pneumopathie inflammatoire ou les patients atteints d'insuffisance rénale modérée ou sévère peuvent présenter un risque accru de développer une PID/pneumopathie inflammatoire et doivent être étroitement surveillés (voir rubrique Posologie et mode d'administration).
Neutropénie
Des cas de neutropénie, y compris de neutropénie fébrile d'issue fatale, ont été rapportés au cours des études cliniques d'Enhertu. Les valeurs de l'hémogramme doivent être contrôlées avant l'instauration du traitement par Enhertu et avant chaque perfusion, et lorsque le tableau clinique le justifie. Selon la sévérité de la neutropénie, une interruption du traitement par Enhertu ou une réduction de la dose peut être nécessaire (voir rubrique Posologie et mode d'administration).
Dysfonction ventriculaire gauche
Des cas de diminution de la fraction d'éjection ventriculaire gauche (FEVG) ont été observés avec les traitements anti-HER2. Un examen standard de la fonction cardiaque (échocardiographie ou ventriculographie isotopique) doit être réalisé pour évaluer la FEVG avant l'instauration du traitement par Enhertu et à intervalles réguliers pendant le traitement si le tableau clinique le justifie. La prise en charge de la diminution de la FEVG doit consister en une interruption du traitement. Le traitement par Enhertu doit être arrêté définitivement en cas de confirmation de FEVG < 40 % ou de diminution absolue > 20 % par rapport à la valeur initiale. Le traitement par Enhertu doit être arrêté définitivement chez les patients présentant une insuffisance cardiaque congestive (ICC) symptomatique (voir le tableau 2 à la rubrique Posologie et mode d'administration).
Toxicité embryonnaire et fœtale
Enhertu peut avoir des effets délétères chez le fœtus lorsqu'il est administré à une femme enceinte. Depuis la commercialisation du trastuzumab, un antagoniste des récepteurs HER2, des cas d'oligohydramnios se manifestant par une hypoplasie pulmonaire fatale, des anomalies du squelette et une mortalité néonatale ont été rapportés lors de son utilisation pendant la grossesse. Sur la base des observations chez l'animal et du mécanisme d'action du DXd, le composant d'Enhertu inhibiteur de la topoisomérase I, celui-ci peut également provoquer une toxicité embryonnaire et fœtale en cas d'administration à une femme enceinte (voir rubrique Fertilité, grossesse et allaitement).
Chez les femmes en âge de procréer, la présence éventuelle d'une grossesse doit être vérifiée avant l'instauration du traitement par Enhertu. La patiente doit être informée des risques potentiels pour le fœtus. Les femmes en âge de procréer doivent être informées qu'elles doivent utiliser une contraception efficace pendant le traitement et pendant au moins 7 mois après la dernière perfusion d'Enhertu. Les hommes ayant une partenaire en âge de procréer doivent être informés qu'ils doivent utiliser une contraception efficace pendant le traitement et pendant au moins 4 mois après la dernière perfusion d'Enhertu (voir rubrique Fertilité, grossesse et allaitement).
Insuffisance hépatique modérée ou sévère
Les données chez les patients présentant une insuffisance hépatique modérée sont limitées et il n'existe pas de données chez les patients atteints d'insuffisance hépatique sévère. Le métabolisme et l'excrétion hépatobiliaire étant les principales voies d'élimination du DXd, l'inhibiteur de la topoisomérase I, Enhertu doit être administré avec prudence chez les patients présentant une insuffisance hépatique modérée ou sévère (voir rubriques Posologie et mode d'administration et Propriétés pharmacocinétiques).
Résumé du profil de sécurité
Enhertu 5,4 mg/kg
La
population globale d'analyse de la sécurité était composée des patients ayant
reçu au moins une dose d'Enhertu 5,4 mg/kg (n = 2 335) pour différents types de
tumeurs dans les études cliniques. La durée
de traitement médiane
dans cette population globale était de 9,0 mois (plage : 0,7 à 45,1 mois).
Les effets indésirables les plus fréquents étaient : nausées (71,1 %), fatigue (55,3 %), vomissements (37,3 %), alopécie (36,1 %), anémie (35,9 %), neutropénie (35,1 %), constipation (31,7 %), appétitdiminué (30,6 %), diarrhée (30,1 %), transaminases augmentées (26,6 %), douleur musculosquelettique (23,6 %), thrombopénie (23,1 %) et leucopénie (21,5 %).
Les effets indésirables de grade 3 ou 4 des Critères communs de terminologie pour les événements indésirables du National Cancer Institute (NCI-CTCAE v.5.0) étaient : neutropénie (18,0 %), anémie (10,5 %), fatigue (7,8 %), leucopénie (6,0 %), thrombopénie (5,4 %), nausées (4,9 %), lymphopénie(3,9 %), hypokaliémie (3,8 %), transaminases augmentées (3,5 %), diarrhée (2,5 %), vomissements (2,4 %), appétit diminué (1,8 %), pneumonie (1,3 %) et fraction d'éjection diminuée (1,0 %). Des effets indésirables de grade 5 sont survenus chez 1,4 % des patients, dont une PID/pneumopathie inflammatoire chez 1,1 % des patients.
Des interruptions du traitement en raison d'effets indésirables ont été rapportées chez 32,6 % des patients traités par Enhertu. Les effets indésirables les plus fréquents ayant entraîné l'interruption du traitement étaient : neutropénie (12,4 %), fatigue (4,7 %), anémie (4,6 %), leucopénie (3,2 %), infection des voies aériennes supérieures (3,0 %), PID/pneumopathie inflammatoire (2,6 %), thrombopénie (2,4 %) et pneumonie (2,0 %). Des réductions de dose ont été rapportées chez 20,3 % des patients traités par Enhertu. Les effets indésirables les plus fréquents ayant entraîné une réduction de dose étaient : fatigue (5,1 %), nausées (4,8 %) neutropénie (3,5 %) et thrombopénie (2,3 %). Le traitement par Enhertu a été arrêté en raison d'un effet indésirable chez 11,7 % des patients. La pneumopathie interstitielle diffuse/pneumopathie inflammatoire était l'effet indésirable le plus fréquent ayant entraîné l'arrêt définitif du traitement (8,4 %).
Enhertu 6,4 mg/kg
La
population globale d'analyse de la sécurité était composée des patients ayant
reçu au moins une dose d'Enhertu 6,4 mg/kg (n = 669) pour différents types de
tumeurs dans les études cliniques. La durée de traitement médiane dans cette population globale
était de 5,7 mois (plage
: 0,7 à 41,0 mois).
Les effets indésirables les plus fréquents étaient : nausées (72,2 %), fatigue (58,4 %), appétit diminué (53,5 %), anémie (44,7 %), neutropénie (43,5 %), vomissements (40,1 %), diarrhée (35,9 %), alopécie(35,4 %), constipation (32,3 %), thrombopénie (30,8 %), leucopénie (29,3 %) et transaminasesaugmentées (24,2 %).
Les effets indésirables de grade 3 ou 4 des Critères communs de terminologie pour les événements indésirables du National Cancer Institute (NCI-CTCAE v.5.0) étaient : neutropénie (28,7 %), anémie (22,6 %), leucopénie (13,3 %), thrombopénie (9,1 %), fatigue (8,4 %), appétit diminué (7,8 %),lymphopénie (6,9 %), nausées (5,8 %), transaminases augmentées (4,3 %), hypokaliémie (4,3 %), pneumonie (3,1 %), neutropénie fébrile (2,8 %), vomissements (2,4 %), diarrhée (2,2 %), poids diminué (1,9 %), phosphatase alcaline sanguine augmentée (1,6 %), pneumopathie interstitielle diffuse (PID, 1,5 %), dyspnée (1,2 %), fraction d'éjection diminuée (1,2 %) et bilirubine sanguine augmentée (1,2 %). Des effets indésirables de grade 5 sont survenus chez 2,7 % of patients, dont une PID chez 2,1 % des patients.
Des interruptions du traitement en raison d'effets indésirables ont été rapportées chez 40,7 % des patients traités par Enhertu. Les effets indésirables les plus fréquents ayant entraîné l'interruption du traitement étaient : neutropénie (16,6 %), anémie (7,8 %), fatigue (5,7 %), PID (4,8 %), leucopénie (4,2 %), appétit diminué (3,7 %), pneumonie (3,6 %), infection des voies aériennes supérieures(3,4 %) et thrombopénie (3,1 %). Des réductions de dose ont été rapportées chez 31,1 % des patients traités par Enhertu. Les effets indésirables les plus fréquents ayant entraîné une réduction de dose étaient : fatigue (10,6 %), neutropénie (6,6 %), nausées (6,4 %), appétit diminué (5,4 %) et thrombopénie (3,0 %). Le traitement par Enhertu a été arrêté en raison d'un effet indésirable chez 17,6 % des patients. La pneumopathie interstitielle diffuse était l'effet indésirable le plus fréquent ayant entraîné l'arrêt définitif du traitement (12,9 %).
Chez les patients présentant un cancer de l'estomac traités par Enhertu 6,4 mg/kg (n = 229), 25,3 % ont reçu une transfusion dans les 28 jours suivant l'apparition d'une anémie ou d'une thrombopénie. Les transfusions étaient administrées principalement pour une anémie.
Liste tabulée des effets indésirables
Les effets indésirables rapportés chez les patients ayant reçu au moins une dose d'Enhertu dans les études cliniques sont présentés dans le tableau 3. Les effets indésirables sont présentés par classe de systèmes d'organes (SOC) et catégories de fréquence MedDRA. Les catégories de fréquence sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000,< 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 3 : Effets indésirables rapportés chez les patients traités par le trastuzumab déruxtécan 5,4 mg/kg ou 6,4 mg/kg pour différents types de tumeurs
|
Classe de systèmes d'organes Catégorie de fréquence |
5,4 mg/kg
Effets indésirables |
6,4 mg/kg
Effets indésirables |
|
Infections et infestations |
||
|
Très fréquent |
Infection des voies aériennes supérieuresa |
Pneumonie, infection des voies aériennes supérieuresa |
|
Fréquent |
Pneumonie |
|
|
Affections hématologiques et du système lymphatique |
||
|
Très fréquent |
Anémieb, neutropéniec, thrombopénied, leucopéniee |
Anémieb, neutropéniec, thrombopénied, leucopéniee |
|
Fréquent |
Lymphopénief, neutropénie fébrile, pancytopénieg |
Neutropénie fébrile, pancytopénieg |
|
Troubles du métabolisme et de la nutrition |
||
|
Très fréquent |
Hypokaliémieh, appétit diminué |
Hypokaliémieh, appétit diminué |
|
Fréquent |
Déshydratation |
Déshydratation |
|
Affections du système nerveux |
||
|
Très fréquent |
Céphaléei |
Céphaléei, dysgueusie |
|
Fréquent |
Sensation vertigineuse, dysgueusie |
Sensation vertigineuse |
|
Affections oculaires |
||
|
Fréquent |
Sécheresse oculaire, vision troublej |
Sécheresse oculaire, vision troublej |
|
Affections respiratoires, thoraciques et médiastinales |
||
|
Très fréquent |
Pneumopathie interstitielle diffusek, toux |
Pneumopathie interstitielle diffusek, dyspnée, toux |
|
Fréquent |
Dyspnée, épistaxis |
Épistaxis |
|
Affections gastro-intestinales |
||
|
Très fréquent |
Nausées, vomissements, constipation, diarrhée, douleur abdominalel, stomatitem, dyspepsie |
Nausées, vomissements, diarrhée, constipation, douleur abdominalel, stomatitem |
|
Fréquent |
Distension abdominale, gastrite, flatulences |
Dyspepsie, distension abdominale, gastrite, flatulences |
|
Affections hépatobiliaires |
||
|
Très fréquent |
Transaminases augmentéesn |
Transaminases augmentéesn |
|
Affections de la peau et du tissu sous-cutané |
||
|
Très fréquent |
Alopécie |
Alopécie |
|
Fréquent |
Éruption cutanéeo, prurit, hyperpigmentation cutanéep |
Éruption cutanéeo, prurit, hyperpigmentation cutanéep |
|
Affections musculosquelettiques et du tissu conjonctif |
||
|
Très fréquent |
Douleur musculosquelettiqueq |
Douleur musculosquelettiqueq |
|
Troubles généraux et anomalies au site d'administration |
||
|
Très fréquent |
Fatiguer, pyrexie |
Fatiguer, pyrexie, œdème périphérique |
|
Fréquent |
Œdème périphérique |
|
|
Investigations |
||
|
Très fréquent |
Fraction d'éjection diminuées, poids diminué |
Fraction d'éjection diminuées, poids diminué |
|
Fréquent |
Phosphatase alcaline sanguine augmentée, bilirubine sanguine augmentéet, créatine sanguine augmentée |
Phosphatase alcaline sanguine augmentée, bilirubine sanguine augmentéet, créatine sanguine augmentée |
|
Lésions, intoxications et complications liées aux procédures |
||
|
Fréquent |
Réactions liées à la perfusionu |
Réactions liées à la perfusionu |
a Le terme inclut
grippe, syndrome grippal,
rhinopharyngite, pharyngite, sinusite,
rhinite, laryngite et infection des voies aériennes supérieures.
b Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut anémie, hémoglobine diminuée, globules rouges diminués et hématocrite diminué. Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut anémie, hémoglobine diminuée et globules rouges diminués.
c Le terme inclut neutropénie et neutrophiles diminués.
d Le terme inclut thrombopénie et numération plaquettaire diminuée.
e Le terme inclut leucopénie et globules blancs diminués.
f Le terme inclut lymphopénie et numération de lymphocytes diminuée.
g La pancytopénie était définie par la présence des trois critères suivants chez un patient : taux d'hémoglobine < 100 g/L et grade CTCAE ≥ 2, taux de neutrophiles < 1,5 × 109/L et grade CTCAE ≥ 1 et taux de plaquettes < 100 × 109/L sans grade CTCAE manquant, selon les résultats des analyses des échantillons prélevés à la même date et/ou selon les critères relatifs au terme préférentiel « pancytopénie ».
h Le terme inclut hypokaliémie et potassium sanguin diminué.
j Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut céphalées, céphalée d'origine sinusienne et migraine. Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut céphalée et migraine.
j Le terme inclut vision trouble et défauts visuels.
k Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut les événements ayant été évalués comme constituant une pneumopathie interstitielle diffuse : insuffisance respiratoire aiguë (n = 2), alvéolite (n = 2), bronchectasie (n = 1), progression de la maladie (n = 1), pneumopathie d'hypersensibilité(n = 1), pneumonie interstitielle idiopathique (n = 1), pneumopathie interstitielle diffuse (n = 109), infection des voies respiratoires inférieures (n = 1), trouble pulmonaire (n = 1), infiltration pulmonaire (n = 1), opacité pulmonaire (n = 4), lymphangite (n = 1), pneumonie organisée (n = 9), pneumonie (n = 9), pneumonie bactérienne (n = 2), pneumonie fongique (n = 1), pneumopathie inflammatoire (n = 136), fibrose pulmonaire (n = 2), masse pulmonaire (n = 1), toxicité pulmonaire (n = 3), pneumopathie radique (n = 4), insuffisance respiratoire (n = 5). Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut les événements ayant été évalués comme constituant une PID liée au traitement : pneumopathie inflammatoire (n = 75), pneumopathie interstitielle diffuse (n = 39), pneumonie organisée (n = 4), insuffisance respiratoire (n = 4), opacité pulmonaire (n = 2), pneumonie (n = 1) et pneumopathie radique (n = 1).
l Le terme inclut gêne abdominale, douleurs gastro-intestinales, douleurs abdominales, douleur abdominale basse et douleur abdominale haute.
m Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut stomatite, ulcère aphteux, ulcération buccale, érosion de la muqueuse buccale et éruption buccale. Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut stomatite uniquement.
n Le terme inclut transaminases augmentées, alanine aminotransférase augmentée, aspartate aminotransférase augmentée, gamma-glutamyltransférase augmentée, fonction hépatique anormale, anomalies du bilan hépatique, augmentation des paramètres hépatiques et hypertransaminasémie.
o Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut éruption cutanée, éruption cutanée pustuleuse, éruption cutanée maculopapuleuse, éruption cutanée papuleuse, éruption cutanée maculeuse et éruption cutanée prurigineuse. Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut éruption cutanée, éruption cutanée pustuleuse, éruption cutanée maculopapuleuse et éruption cutanée prurigineuse.
p Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut hyperpigmentation cutanée, modification de la couleur de la peau et trouble de la pigmentation. Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut hyperpigmentation cutanée et trouble de la pigmentation.
q Le terme inclut dorsalgie, myalgie, douleur dans les extrémités, douleur musculosquelettique, contractures musculaires, douleur osseuse, cervicalgie, douleur musculosquelettique du thorax et gêne dans un membre.
r Le terme inclut asthénie, fatigue, malaise et léthargie.
s Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme « diminution de la fraction d'éjection » inclut les paramètres paracliniques de diminution de la FEVG (n = 312) et/ou les termes préférentiels fraction d'éjection diminuée (n = 99), insuffisance cardiaque (n = 5), insuffisance cardiaque aiguë (n = 1), insuffisance cardiaque chronique (n = 1), insuffisance cardiaque congestive (n = 1) et dysfonction ventriculaire gauche (n = 3). Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme« diminution de la fraction d'éjection » inclut les paramètres paracliniques de diminution de la FEVG (n = 97) et/ou les termes préférentiels fraction d'éjection diminuée (n = 11) et dysfonction ventriculaire gauche (n = 1).
t Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut bilirubine sanguine augmentée, hyperbilirubinémie, bilirubine conjuguée augmentée et bilirubine non conjuguée augmentée. Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut bilirubine sanguine augmentée, hyperbilirubinémie et bilirubine conjuguée augmentée.
u Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, les cas de réactions liées à la perfusion incluent réaction liée à la perfusion (n = 23) et hypersensibilité (n = 2). Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, les cas de réactions liées à la perfusion incluent réaction liée à la perfusion(n = 6) et hypersensibilité (n = 1). Tous les cas de réactions liées à la perfusion étaient de grades 1 et 2.
b Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut anémie, hémoglobine diminuée, globules rouges diminués et hématocrite diminué. Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut anémie, hémoglobine diminuée et globules rouges diminués.
c Le terme inclut neutropénie et neutrophiles diminués.
d Le terme inclut thrombopénie et numération plaquettaire diminuée.
e Le terme inclut leucopénie et globules blancs diminués.
f Le terme inclut lymphopénie et numération de lymphocytes diminuée.
g La pancytopénie était définie par la présence des trois critères suivants chez un patient : taux d'hémoglobine < 100 g/L et grade CTCAE ≥ 2, taux de neutrophiles < 1,5 × 109/L et grade CTCAE ≥ 1 et taux de plaquettes < 100 × 109/L sans grade CTCAE manquant, selon les résultats des analyses des échantillons prélevés à la même date et/ou selon les critères relatifs au terme préférentiel « pancytopénie ».
h Le terme inclut hypokaliémie et potassium sanguin diminué.
j Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut céphalées, céphalée d'origine sinusienne et migraine. Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut céphalée et migraine.
j Le terme inclut vision trouble et défauts visuels.
k Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut les événements ayant été évalués comme constituant une pneumopathie interstitielle diffuse : insuffisance respiratoire aiguë (n = 2), alvéolite (n = 2), bronchectasie (n = 1), progression de la maladie (n = 1), pneumopathie d'hypersensibilité(n = 1), pneumonie interstitielle idiopathique (n = 1), pneumopathie interstitielle diffuse (n = 109), infection des voies respiratoires inférieures (n = 1), trouble pulmonaire (n = 1), infiltration pulmonaire (n = 1), opacité pulmonaire (n = 4), lymphangite (n = 1), pneumonie organisée (n = 9), pneumonie (n = 9), pneumonie bactérienne (n = 2), pneumonie fongique (n = 1), pneumopathie inflammatoire (n = 136), fibrose pulmonaire (n = 2), masse pulmonaire (n = 1), toxicité pulmonaire (n = 3), pneumopathie radique (n = 4), insuffisance respiratoire (n = 5). Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut les événements ayant été évalués comme constituant une PID liée au traitement : pneumopathie inflammatoire (n = 75), pneumopathie interstitielle diffuse (n = 39), pneumonie organisée (n = 4), insuffisance respiratoire (n = 4), opacité pulmonaire (n = 2), pneumonie (n = 1) et pneumopathie radique (n = 1).
l Le terme inclut gêne abdominale, douleurs gastro-intestinales, douleurs abdominales, douleur abdominale basse et douleur abdominale haute.
m Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut stomatite, ulcère aphteux, ulcération buccale, érosion de la muqueuse buccale et éruption buccale. Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut stomatite uniquement.
n Le terme inclut transaminases augmentées, alanine aminotransférase augmentée, aspartate aminotransférase augmentée, gamma-glutamyltransférase augmentée, fonction hépatique anormale, anomalies du bilan hépatique, augmentation des paramètres hépatiques et hypertransaminasémie.
o Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut éruption cutanée, éruption cutanée pustuleuse, éruption cutanée maculopapuleuse, éruption cutanée papuleuse, éruption cutanée maculeuse et éruption cutanée prurigineuse. Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut éruption cutanée, éruption cutanée pustuleuse, éruption cutanée maculopapuleuse et éruption cutanée prurigineuse.
p Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut hyperpigmentation cutanée, modification de la couleur de la peau et trouble de la pigmentation. Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut hyperpigmentation cutanée et trouble de la pigmentation.
q Le terme inclut dorsalgie, myalgie, douleur dans les extrémités, douleur musculosquelettique, contractures musculaires, douleur osseuse, cervicalgie, douleur musculosquelettique du thorax et gêne dans un membre.
r Le terme inclut asthénie, fatigue, malaise et léthargie.
s Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme « diminution de la fraction d'éjection » inclut les paramètres paracliniques de diminution de la FEVG (n = 312) et/ou les termes préférentiels fraction d'éjection diminuée (n = 99), insuffisance cardiaque (n = 5), insuffisance cardiaque aiguë (n = 1), insuffisance cardiaque chronique (n = 1), insuffisance cardiaque congestive (n = 1) et dysfonction ventriculaire gauche (n = 3). Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme« diminution de la fraction d'éjection » inclut les paramètres paracliniques de diminution de la FEVG (n = 97) et/ou les termes préférentiels fraction d'éjection diminuée (n = 11) et dysfonction ventriculaire gauche (n = 1).
t Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, le terme inclut bilirubine sanguine augmentée, hyperbilirubinémie, bilirubine conjuguée augmentée et bilirubine non conjuguée augmentée. Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, le terme inclut bilirubine sanguine augmentée, hyperbilirubinémie et bilirubine conjuguée augmentée.
u Dans le traitement de tous les types de tumeurs à la dose de 5,4 mg/kg, les cas de réactions liées à la perfusion incluent réaction liée à la perfusion (n = 23) et hypersensibilité (n = 2). Dans le traitement de tous les types de tumeurs à la dose de 6,4 mg/kg, les cas de réactions liées à la perfusion incluent réaction liée à la perfusion(n = 6) et hypersensibilité (n = 1). Tous les cas de réactions liées à la perfusion étaient de grades 1 et 2.
Description de certains effets indésirables
Pneumopathie interstitielle diffuse/pneumopathie inflammatoire
Chez les
patients traités par Enhertu 5,4 mg/kg pour différents types de tumeurs dans
les études cliniques (n = 2 335), des cas de pneumopathie interstitielle
diffuse (PID), pneumopathie inflammatoire, pneumonie organisée et pneumopathie
interstitielle aiguë ont été observés par les investigateurs chez 13,3 % des
patients. La PID/pneumopathie inflammatoire a été confirmée par revue
indépendante chez 12,2 % des patients, elle a entraîné l'arrêt du traitement
chez 8,4 % des patients et l'interruption du traitement chez 2,6 % des
patients. Dans la majorité des cas, la PID/pneumopathie inflammatoire était de
grade 1 (2,9 %) ou de grade 2 (7,5 %). Des cas de grade 3 sont survenus chez 0,7
% des patients et un cas de grade
4 a été rapporté. Des événements de grade 5 (d'issue fatale) sont survenus chez 1,1 % des
patients. Le délai médian d'apparition était de 5,5 mois (plage : -0,3 à 31,5
mois) en incluant une PID préexistante confirmée par revue indépendante chez
deux patients. Après un suivi
médian de 280 jours, il n'a pas été observé
de récupération chez 30,8 % des patients présentant une PID/pneumopathie inflammatoire confirmée par revue
indépendante (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Chez les patients traités par Enhertu 6,4 mg/kg pour différents types de tumeurs dans les études cliniques (n = 669), une PID est survenue chez 17,9 % des patients. Dans la majorité des cas, la PID était de grade 1 (4,9 %) ou de grade 2 (9,4 %). Des cas de grade 3 sont survenus chez 1,3 % des patients et des cas de grade 4 sont survenus chez 0,1 % des patients. Des événements de grade 5 (d'issue fatale) sont survenus chez 2,1 % des patients. Un patient présentait une PID préexistante quis'est aggravée après le traitement et a évolué en PID de grade 5 (d'issue fatale). Le délai médian d'apparition était de 4,2 mois (plage : -0,5 à 21,0 mois) (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Neutropénie
Chez les
patients traités par Enhertu 5,4 mg/kg pour différents types de tumeurs dans
les études cliniques (n = 2 335), une neutropénie a été rapportée chez 35,1 %
des patients et les événements étaient de grade
3 ou 4 chez 18,0 % des patients. Le délai médian
d'apparition était de 42
jours (plage : 1 jour à 31,9
mois) et la durée médiane du premier événement était de 21 jours (plage : 1
jour à 17,1 mois). Une neutropénie fébrile a été rapportée chez 1,0
% des patients et < 0,1 % des cas ont été de grade 5 (voir rubrique Posologie et mode d'administration).
Chez les patients traités par Enhertu 6,4 mg/kg pour différents types de tumeurs dans les études cliniques (n = 669), une neutropénie a été rapportée chez 43,5 % des patients et les événements étaient de grade 3 ou 4 chez 28,7 % des patients. Le délai médian d'apparition était de 16 jours (plage : 1 jour à 24,8 mois) et la durée médiane du premier événement était de 9 jours (plage : 2 jours à 17,2 mois). Une neutropénie fébrile a été rapportée chez 3,0 % des patients et 0,1 % des cas ont été de grade 5 (voir rubrique Posologie et mode d'administration).
Dysfonction ventriculaire gauche
Chez les
patients traités par Enhertu 5,4 mg/kg pour différents types de tumeurs dans
les études cliniques (n = 2 335), une diminution de la FEVG a été rapportée
chez 108 patients (4,6 %), dont 14 cas de grade 1 (0,6 %), 80 cas de grade 2 (3,4 %), 13 cas de grade 3 (0,6 %) et
1 cas de grade 4(< 0,1 %). La fréquence observée de
diminution de la FEVG sur la base des paramètres paracliniques
(échocardiographie ou ventriculographie isotopique) était de 296/2
075 (14,3 %) pour les événements
de grade 2 et de 15/2 075 (0,7 %) pour les événements de grade 3. Enhertu n'a pas été étudié
chez les patients ayant une FEVG inférieure à 50 % avant l'instauration du
traitement (voir rubrique Posologie et mode d'administration).
La dysfonction ventriculaire gauche a entraîné l'interruption du traitement chez 1,2 % des patients (27/2 335). Le délai médian d'apparition de la diminution de la FEVG de grade le plus élevé était de 4,8 mois et le délai médian de récupération (FEVG ≥ 90 % de la valeur initiale) par rapport à la diminution de la FEVG de grade le plus élevé était de 6,3 mois.
Chez les patients traités par Enhertu 6,4 mg/kg pour différents types de tumeurs dans les études cliniques (n = 669), une diminution de la FEVG a été rapportée chez 12 patients (1,8 %), dont 1 cas de grade 1 (0,1 %), 8 cas de grade 2 (1,2 %) et 3 cas de grade 3 (0,4 %). La fréquence observée de diminution de la FEVG sur la base des paramètres paracliniques (échocardiographie ou ventriculographie isotopique) était de 89/597 (14,9 %) pour les événements de grade 2 et de 8/597 (1,3 %) pour les événements de grade 3.
Réactions à la perfusion
Chez les patients traités par Enhertu 5,4 mg/kg pour différents types de tumeurs dans les études cliniques (n = 2 335), des réactions à la perfusion ont été rapportées chez 25 patients (1,1 %), la majorité des cas étant de grade 1 ou de grade 2. Cinq événements (0,2 %) de réaction à la perfusion ont entraîné l'interruption du traitement et un événement (< 0,1 %) a entraîné l'arrêt du traitement.
Chez les patients traités par Enhertu 6,4 mg/kg pour différents types de tumeurs dans les études cliniques (n = 669), des réactions à la perfusion ont été rapportées chez 7 patients (1,0 %), tous les cas étant de grade 1 ou de grade 2. Il n'a pas été rapporté d'événements de grade 3. Un événement (0,1 %) de réaction à la perfusion a entraîné l'interruption du traitement et aucun événement n'a entraîné l'arrêt du traitement.
Immunogénicité
Comme avec toutes les protéines thérapeutiques, il existe une possibilité d'immunogénicité. Pour les doses de 5,4 mg/kg et 6,4 mg/kg testées dans les études cliniques, 2,2 % des patients évaluables (70/3 124) avaient développé des anticorps contre le trastuzumab déruxtécan après le traitement parEnhertu. L'incidence d'anticorps neutralisants dirigés contre le trastuzumab déruxtécan apparus sous traitement était de 0,1 % (3/3 124). Il n'a pas été observé d'effet apparent entre le développement d'anticorps et la pharmacocinétique, la sécurité et/ou l'efficacité d'Enhertu.
Population pédiatrique
La sécurité n'a pas été établie dans cette population.
Sujets âgés
Chez les patients traités par Enhertu 5,4 mg/kg pour différents types de tumeurs dans les études cliniques (n = 2 335), 28,9 % étaient âgés de 65 ans et plus et 6,3 % étaient âgés de 75 ans et plus. Il a été observé une incidence supérieure d'effets indésirables de grades 3 et 4 chez les patients âgés de 65 ans et plus (48,4 %) par rapport aux patients de moins de 65 ans (43,2 %), ce qui a entraîné un taux plus élevé d'arrêts du traitement en raison d'effets indésirables. L'incidence d'effets indésirables d'issue fatale était de 2,4 % chez les patients âgés de 65 ans et plus et de 1 % chez les patients âgés de moins de 65 ans.
Sur les
669 patients traités par Enhertu 6,4 mg/kg pour différents types de tumeurs
dans les études cliniques, 39,2 % étaient âgés de 65 ans et plus et 7,6 %
étaient âgés de 75 ans et plus. L'incidence d'effets indésirables de grades 3
et 4 observée chez les patients âgés de 65 ans et plus était de 59,9 % contre
62,9 % chez les patients plus jeunes. Il a été observé une incidence supérieure
d'effets indésirables de grades 3 et 4 chez les patients âgés de 75 ans et plus
(64,7 %) par rapport aux patients de moins de
75 ans (61,5 %). Chez les patients âgés de 75 ans et plus,
l'incidence d'effets indésirables graves (37,3 %) et d'événements d'issue
fatale (7,8 %) était plus élevée que chez les patients de moins
de 75 ans (20,7 % et 2,3 %). Les données permettant d'établir la sécurité chez
les patients âgés de 75 ans et plus sont limitées.
Différences ethniques
Dans les études cliniques, il n'a pas été observé de différences pertinentes de l'exposition ou de l'efficacité entre les patients de différents groupes ethniques. Chez les patients asiatiques traités par Enhertu 6,4 mg/kg, il a été observé une incidence plus élevée (différence ≥ 10 %) de neutropénie (58,1 % versus 18,6 %), d'anémie (51,1 % versus 32,4 %), de leucopénie (42,7 % versus 6,9 %), de thrombopénie (40,5 % versus 15,4 %) et de lymphopénie (17,6 % versus 7,3 %) que chez les patients d'autres groupes ethniques. Chez les patients asiatiques, 4,3 % ont présenté un événement de saignement dans les 14 jours suivant l'apparition d'une thrombopénie, contre 1,6 % des patients d'autres groupes ethniques.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
NE PAS ADMINISTRER en
injection intraveineuse rapide ou en bolus.
S'ASSURER du statut tumoral HER2 ou de la présence de la mutation activatrice
du gène en fonction du type de cancer :
Cancer du sein HER2-positif
Les patients traités par le trastuzumab déruxtécan pour un cancer du sein
doivent avoir un statut tumoral HER2-positif documenté, défini comme un score
3+ par immunohistochimie (IHC) ou comme un ratio ≥ 2 par hybridation in situ
(HIS) ou hybridation in situ en fluorescence (FISH), déterminés en
utilisant un dispositif médical de diagnostic in vitro (DIV) portant le
marquage CE. Si un DIV portant le marquage CE n'est pas disponible, le statut
HER2 doit être déterminé à l'aide d'un autre test validé.
Cancer du sein HER2-faible ou ultrafaible
Les patients traités par le trastuzumab déruxtécan doivent avoir
un statut tumoral HER2-faible documenté, défini comme un score IHC 1+ ou IHC
2+/HIS-, ou un statut tumoral HER2-ultrafaible, établi par un score IHC 0 avec
marquage membranaire (IHC > 0 < 1+), déterminés en utilisant un DIV
portant le marquage CE. Si un DIV portant le marquage CE n'est pas disponible,
le statut HER2 doit être déterminé à l'aide d'un autre test validé.
CBNPC
Les patients traités par le trastuzumab déruxtécan pour un CBNPC de stade
avancé doivent être porteurs d'une mutation activatrice du gène HER2 (ERBB2),
détectée en utilisant un dispositif médical de diagnostic in vitro (DIV)
portant le marquage CE. Si un DIV portant le marquage CE n'est pas disponible,
le statut mutationnel d'HER2 doit être déterminé à l'aide d'un autre test
validé.
Cancer de l'estomac
Les patients traités par le trastuzumab déruxtécan pour un cancer de l'estomac
ou de la jonction oesogastrique doivent avoir un statut tumoral HER2-positif
documenté, défini comme un score 3+ par immunohistochimie (IHC) ou comme un
ratio ≥ 2 par hybridation in situ (HIS) ou hybridation in situ en
fluorescence (FISH), déterminés en utilisant un dispositif médical de
diagnostic in vitro (DIV) portant le marquage CE. Si un DIV portant le
marquage CE n'est pas disponible, le statut HER2 doit être déterminé à l'aide
d'un autre test validé.
Prémédication
Avant l'administration de chaque dose, les patients doivent recevoir
une prémédication consistant en une association de deux ou trois
médicaments (par exemple dexaméthasone avec un antagoniste des
récepteurs 5-HT3 et/ou un antagoniste des récepteurs NK1, ainsi que
d'autres médicaments s'ils sont indiqués) pour la prévention des
nausées et vomissements chimio-induits.
SURVEILLANCE DU TRAITEMENT :
- Hémogramme AVANT l'instauration du traitement, avant chaque perfusion, et
lorsque le tableau clinique le justifie.
- Examen standard de la fonction cardiaque (échocardiographie ou
ventriculographie isotopique) pour évaluer la FEVG AVANT l'instauration du
traitement et à intervalles réguliers PENDANT le traitement si le tableau
clinique le justifie.
- Signes et symptômes de pneumopathie interstitielle diffuse/pneumopathie inflammatoire.
FEMMES EN AGE DE PROCRÉER :
- VÉRIFIER la présence éventuelle d'une grossesse avant l'instauration du
traitement.
- INFORMER des risques potentiels pour le foetus en cas de grossesse.
- Mettre en place une contraception efficace pendant le traitement et au
moins sept mois après l'arrêt du traitement.
HOMMES :
- ayant une partenaire en âge de procréer : Utiliser une contraception efficace
pendant le traitement et au moins quatre mois après l'arrêt du traitement.
- Conseiller la conservation du sperme AVANT l'instauration du traitement.
- Conseiller aux patients de ne pas faire de don de sperme, ni demander de
congélation du sperme PENDANT toute la période de traitement et au moins quatre
mois après la fin du traitement.
ALLAITEMENT et NOUVEAU-NE
de mère traitée :
- Ne pas allaiter pendant le traitement par le trastuzumab déruxtécan ou
pendant sept mois après la fin du traitement.
- SURVEILLANCE étroite du nouveau-né en cas de grossesse pendant le traitement
ou dans les sept mois qui suivent la fin du traitement.
Femmes en âge de procréer/Contraception chez les hommes et les femmes
Chez les femmes en âge de procréer, la présence éventuelle d'une grossesse doit être vérifiée avant l'instauration du traitement par Enhertu.
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par Enhertu et pendant au moins sept mois après l'arrêt du traitement.
Les hommes ayant une partenaire en âge de procréer doivent utiliser une contraception efficace pendant le traitement par Enhertu et pendant au moins quatre mois après l'arrêt du traitement.
Grossesse
Il n'existe pas de données sur l'utilisation d'Enhertu chez la femme enceinte. Cependant, le trastuzumab, un antagoniste des récepteurs HER2, peut avoir des effets délétères chez le fœtus lorsqu'il est administré à une femme enceinte. Depuis la commercialisation du trastuzumab, des cas d'oligohydramnios se manifestant parfois par une hypoplasie pulmonaire fatale, des anomalies du squelette et une mortalité néonatale ont été rapportés lors de son utilisation pendant la grossesse. Sur la base des observations chez l'animal et du mécanisme d'action du DXd, le composant d'Enhertu inhibiteur de la topoisomérase I, une toxicité embryonnaire et fœtale est prévisible en cas d'administration à une femme enceinte (voir rubrique Données de sécurité préclinique).
L'administration d'Enhertu chez les femmes enceintes n'est pas recommandée et les patientes doivent être informées des risques potentiels pour le fœtus avant qu'elles ne deviennent enceintes. Les patientes qui découvrent qu'elles sont enceintes doivent contacter immédiatement leur médecin. En cas de grossesse survenant pendant le traitement par Enhertu ou dans les sept mois suivant la fin du traitement, une surveillance étroite est recommandée.
Allaitement
On ne sait pas si le trastuzumab déruxtécan est excrété dans le lait maternel. Les IgG humaines sont secrétées dans le lait maternel et la possibilité d'absorption et d'effets indésirables graves chez le nourrisson n'est pas connue. Par conséquent, les femmes ne doivent pas allaiter pendant le traitement par Enhertu ou pendant sept mois après la fin du traitement. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre le traitement avec Enhertu en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Il n'a pas été réalisé d'études spécifiques de la fertilité avec le trastuzumab déruxtécan. Selon les résultats des études de toxicologie effectuées chez l'animal, Enhertu peut altérer les fonctions de reproduction et la fertilité masculines. On ne sait pas si le trastuzumab déruxtécan ou ses métabolites sont présents dans le liquide séminal. Avant l'instauration du traitement, il doit être recommandé aux patients de se faire conseiller sur la conservation du sperme. Les patients ne doivent pas faire de don de sperme ni demander de congélation du sperme pendant toute la période de traitement par Enhertu et pendant au moins quatre mois après la fin du traitement.
L'administration concomitante avec le ritonavir, un inhibiteur d'OATP1B, du CYP3A et de la P-gp ou avec l'itraconazole, un inhibiteur puissant du CYP3A et de la P-gp, a entraîné une augmentation non cliniquement significative (d'environ 10 à 20 %) des expositions au trastuzumab déruxtécan ou à l'inhibiteur de la topoisomérase I libéré, le DXd. Aucun ajustement de la posologie n'est nécessaire en cas d'administration concomitante de trastuzumab déruxtécan avec des médicaments qui sont des inhibiteurs du CYP3A ou des transporteurs OATP1B ou P-gp (voir rubrique Propriétés pharmacocinétiques).
Enhertu doit être prescrit par un médecin et administré sous la surveillance d'un professionnel de santé expérimenté dans l'utilisation des médicaments anticancéreux. Afin d'éviter des erreurs médicamenteuses, il est important de vérifier l'étiquette du flacon pour s'assurer que le médicament préparé et administré est Enhertu (trastuzumab déruxtécan) et non le trastuzumab ou le trastuzumab emtansine.
Enhertu ne doit pas être remplacé par le trastuzumab ou le trastuzumab emtansine.
Sélection des patients
Cancer du sein HER2-positif
Les patients traités par le trastuzumab déruxtécan pour un cancer du sein doivent avoir un statut
tumoral HER2-positif documenté, défini comme un score 3+ par immunohistochimie
(IHC) ou comme un ratio ≥ 2 par hybridation in situ (HIS) ou
hybridation in situ en fluorescence (FISH), déterminés en utilisant un
dispositif médical de diagnostic in vitro (DIV) portant le marquage CE.
Si un DIV portant le marquage CE n'est pas disponible, le statut HER2 doit être
déterminé à l'aide d'un autre test validé.
Cancer du sein HER2-faible ou HER2-ultrafaible
Les patients traités par le trastuzumab déruxtécan doivent avoir un
statut tumoral HER2-faible documenté, défini comme un score IHC 1+ ou
IHC 2+/HIS-, ou un statut tumoral HER2-ultrafaible, établi par un score
IHC 0 avec marquage membranaire (IHC > 0 < 1+), déterminés en
utilisant un DIV portant le marquage CE. Si un DIV portant le marquage
CE n'est pas disponible, le statut HER2 doit être déterminé à l'aide
d'un autre test validé (voir rubrique Propriétés pharmacodynamiques).
CBNPC
Les patients traités par le trastuzumab déruxtécan pour un CBNPC de stade avancé doivent être
porteurs d'une mutation activatrice du gène HER2 (ERBB2), détectée en utilisant
un dispositif médical de diagnostic in vitro (DIV) portant le marquage
CE. Si un DIV portant le marquage CE n'est pas disponible, le statut
mutationnel d'HER2 doit être déterminé à l'aide d'un autre test validé.
Cancer de l'estomac
Les patients traités par le trastuzumab déruxtécan pour un cancer de l'estomac ou de la jonction
œsogastrique doivent avoir un statut tumoral HER2-positif documenté, défini
comme un score 3+ par immunohistochimie (IHC) ou comme un ratio ≥ 2 par
hybridation in situ (HIS) ou hybridation in situ en fluorescence
(FISH), déterminés en utilisant un dispositif médical de diagnostic in vitro
(DIV) portant le marquage CE. Si un DIV portant le marquage CE n'est pas
disponible, le statut HER2 doit être déterminé à l'aide d'un autre test validé.
Posologie
Cancer du sein
La dose recommandée d'Enhertu est de 5,4 mg/kg,
administrée en perfusion intraveineuse toutes les trois semaines (cycle de 21
jours) jusqu'à la progression de la maladie ou la survenue d'une toxicité
inacceptable.
CBNPC
La dose recommandée d'Enhertu est de 5,4 mg/kg,
administrée en perfusion intraveineuse toutes les trois semaines (cycle de 21
jours) jusqu'à la progression de la maladie ou la survenue d'une toxicité
inacceptable.
Cancer de l'estomac
La dose recommandée d'Enhertu est de 6,4 mg/kg,
administrée en perfusion intraveineuse toutes les trois semaines (cycle de 21
jours) jusqu'à la progression de la maladie ou la survenue d'une toxicité
inacceptable.
La dose initiale doit être administrée en perfusion intraveineuse de 90 minutes. Si la première perfusion a été bien tolérée, les doses suivantes d'Enhertu peuvent être administrées en perfusion de 30 minutes.
Le débit de perfusion doit être ralenti ou la perfusion interrompue si le patient présente des symptômes liés à la perfusion (voir rubrique Effets indésirables). Le traitement par Enhertu doit être arrêté définitivement en cas de réactions sévères à la perfusion.
Prémédication
Enhertu est émétisant (voir rubrique Effets indésirables), ce qui comprend l'induction de nausées et/ou vomissements retardés. Avant l'administration de chaque dose d'Enhertu, les patients doivent recevoir une prémédication consistant en une association de deux ou trois médicaments (par exemple dexaméthasone avec un antagoniste des récepteurs 5-HT3 et/ou un antagoniste des récepteurs NK1, ainsi que d'autres médicaments s'ils sont indiqués) pour la prévention des nausées et vomissements chimio-induits.
Modifications posologiques
La prise en charge des effets indésirables peut nécessiter une interruption temporaire du traitement par Enhertu, une réduction de la dose ou l'arrêt du traitement conformément aux recommandations présentées dans les tableaux 1 et 2.
La dose d'Enhertu ne doit pas être ré-augmentée après qu'une réduction de dose a été effectuée.
Tableau 1 : Schéma de réduction de dose
| Schéma de réduction de dose | Cancer du sein et CBNPC | Cancer de l'estomac |
| Dose initiale recommandée | 5,4 mg/kg | 6,4 mg/kg |
| Première réduction de dose | 4,4 mg/kg | 5,4 mg/kg |
| Deuxième réduction de dose | 3,2 mg/kg | 4,4 mg/kg |
| Autre réduction de dose nécessaire | Arrêter le traitement. | Arrêter le traitement. |
Tableau 2 : Modifications posologiques en cas d'effets indésirables
| Effet indésirable | Intensité | Modification du traitement | |
| Pneumopathie interstitielle diffuse (PID)/pneumopathie inflammatoire | PID/pneumopathie inflammatoire asymptomatique (grade 1) |
Interrompre
le traitement par Enhertu jusqu'à la résolution au
grade 0, puis :
|
|
| PID/pneumopathie inflammatoire symptomatique (grade ≥ 2) |
|
||
| Neutropénie | Grade 3 (0,5 à moins de 1,0 × 109/L) |
|
|
| Grade 4 (moins de 0,5 × 109/L) |
|
||
| Neutropénie fébrile | Nombre absolu de neutrophiles inférieur à 1,0 × 109/L et température > 38,3 °C ou température ≥ 38 °C persistant pendant plus d'une heure |
|
|
| Diminution de la fraction d'éjection ventriculaire gauche (FEVG) | FEVG > 45 % et diminution absolue de 10 % à 20 % par rapport à la valeur initiale |
|
|
| FEVG de 40 % à 45 % | et diminution absolue < 10 % par rapport à la valeur initiale |
|
|
| et diminution absolue de 10 % à 20 % par rapport à la valeur initiale |
|
||
| FEVG < 40 % ou diminution absolue > 20 % par rapport à la valeur initiale |
|
||
| Insuffisance cardiaque congestive (ICC) symptomatique |
|
||
Les grades de toxicité sont définis selon les Critères de terminologie communs pour les événements indésirables du National Cancer Institute version 5.0 (NCI-CTCAE v5.0).
Oubli ou retard de dose
En cas d'oubli ou de retard d'une dose programmée, la dose doit être administrée le plus tôt possible sans attendre le prochain cycle planifié. Le calendrier d'administration doit être ajusté afin de maintenir un intervalle de trois semaines entre les perfusions. La perfusion doit être administrée à la dose et au débit tolérés par le patient lors de la perfusion la plus récente.
Populations particulières
Sujets âgés
Aucun
ajustement de la posologie d'Enhertu n'est nécessaire
chez les patients âgés de 65 ans et plus. Les données disponibles chez les
patients âgés de 75 ans et plus sont limitées.
Insuffisance rénale
Aucun
ajustement de la posologie n'est nécessaire chez les patients présentant une
insuffisance rénale légère (clairance de la créatinine [ClCr]
≥ 60 et < 90 mL/min) ou modérée (ClCr ≥ 30 et < 60 mL/min)
(voir rubrique Propriétés pharmacocinétiques). La nécessité éventuelle
d'un ajustement de la posologie chez les patients présentant une insuffisance
rénale sévère ou une insuffisance rénale terminale ne peut pas être déterminée
car l'insuffisance rénale sévère était un critère de non-inclusion dans les
études cliniques. Une incidence accrue de PID/pneumopathie inflammatoire de
grades 1 et 2 entraînant une augmentation des arrêts du traitement a été
observée chez les patients présentant une insuffisance rénale modérée. Chez les
patients présentant une insuffisance rénale modérée lors de l'inclusion traités
par Enhertu 6,4 mg/kg, il a été observé une incidence
plus élevée d'effets indésirables graves que chez les patients ayant une
fonction rénale normale. Les patients atteints d'insuffisance rénale modérée ou
sévère doivent être étroitement surveillés afin que les effets indésirables
incluant PID/pneumopathie inflammatoire puissent être détectés (voir rubrique Mises
en garde spéciales et précautions d'emploi).
Insuffisance hépatique
Aucun
ajustement de la posologie n'est nécessaire chez les patients ayant un taux de
bilirubine totale ≤ 1,5 fois la limite supérieure de la normale (LSN),
quelle que soit la valeur de l'aspartate aminotransférase
(ASAT). La nécessité éventuelle d'un ajustement de la posologie chez
les
patients ayant un taux de bilirubine totale > 1,5 × LSN, quelle que
soit la
valeur de l'ASAT, ne peut pas être déterminée en raison de données
limitées ; par conséquent, ces patients doivent être étroitement
surveillés
(voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés
pharmacocinétiques).
Population pédiatrique
La
sécurité et l'efficacité d'Enhertu chez les enfants
et adolescents âgés de moins de 18 ans n'ont pas été établies. Aucune donnée
n'est disponible.
Mode d'administration
Voie intraveineuse. Enhertu doit être reconstitué et dilué par un professionnel de santé et administré en perfusion intraveineuse. Il ne doit pas être administré en injection intraveineuse rapide ou en bolus.
Pour les instructions concernant la reconstitution et la dilution du médicament avant administration, voir la rubrique Précautions particulières d'élimination et de manipulation.
Durée de conservation :
Flacon non ouvert
4 ans.
Solution reconstituée
La stabilité physico-chimique après reconstitution a été démontrée pendant une durée allant jusqu'à 48 heures à une température comprise entre 2 °C et 8 °C.
D'un point de vue microbiologique, le produit doit être utilisé immédiatement. Dans le cas contraire, les durées et conditions de conservation après reconstitution et avant utilisation relèvent de la responsabilité de l'utilisateur et ne doivent normalement pas excéder 24 heures à une température comprise entre 2° C et 8° C, sauf si la reconstitution a été effectuée en conditions aseptiques contrôlées et validées.
Solution diluée
Il est recommandé d'utiliser immédiatement la solution diluée. Dans le cas contraire, la solution reconstituée diluée dans une poche à perfusion contenant une solution de glucose à 5 % peut être conservée à température ambiante (≤ 30 ºC) pendant 4 heures au maximum en incluant les durées de préparation et de perfusion ou au réfrigérateur (entre 2 °C et 8 °C) pendant 24 heures au maximum, à l'abri de la lumière.
Précautions particulières de conservation :
À conserver au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler.
Pour les conditions de conservation du médicament après reconstitution et dilution, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments à l'exception de ceux mentionnés dans la rubrique Précautions particulières d'élimination et de manipulation.
Ne pas utiliser de solution injectable de chlorure de sodium pour la reconstitution ou la dilution car cela peut provoquer la formation de particules.
La dose maximale tolérée de trastuzumab déruxtécan n'a pas été établie. Dans les études cliniques, il n'a pas été testé de doses uniques supérieures à 8,0 mg/kg. En cas de surdosage, les patients doivent être étroitement surveillés afin que des signes ou symptômes d'effets indésirables puissent être détectés et un traitement symptomatique approprié doit être instauré.
Classe pharmacothérapeutique : antinéoplasiques, inhibiteurs HER2 (anti-HER2) (récepteur 2 du facteur de croissance épidermique humain), Code ATC : L01FD04.
Mécanisme d'action
Enhertu, trastuzumab déruxtécan, est un anticorps conjugué ciblant le récepteur HER2. L'anticorps est une IgG1 anti-HER2 humanisée couplée au déruxtécan (DXd), un inhibiteur de la topoisomérase I, par un agent de liaison tétrapeptidique clivable. L'anticorps conjugué est stable dans le plasma. La fonction de la composante anticorps est de se lier aux récepteurs HER2 exprimés à la surface de certaines cellules tumorales. Après la liaison, le complexe trastuzumab déruxtécan est alors internalisé et l'agent de liaison est clivé dans la cellule par des enzymes lysosomales dont l'expression est régulée positivement dans les cellules cancéreuses. Une fois libéré, le DXd qui diffuse à travers la membrane provoque des lésions de l'ADN et la mort cellulaire par apoptose. Le DXd, un dérivé de l'exatécan, est environ 10 fois plus puissant que le SN-38, le métabolite actif de l'irinotécan.
Des études in vitro indiquent que la composante anticorps du trastuzumab déruxtécan, qui possède la même séquence d'acides aminés que le trastuzumab, se lie également au récepteur FcγRIIIa et à la fraction C1q du complément. L'anticorps induit une cytotoxicité à médiation cellulaire dépendante des anticorps (ADCC) dans les cellules humaines de cancer du sein qui surexpriment HER2. En outre, l'anticorps inhibe la signalisation par la voie phosphatidyl-inositol-3 kinase (PI3-K) dans les cellules humaines de cancer du sein qui surexpriment HER2.
Efficacité clinique
Cancer du sein HER2-positif
DESTINY-Breast03 (NCT03529110)
L'efficacité et la sécurité d'Enhertu
ont été évaluées
dans l'étude DESTINY-Breast03, une étude dephase III
multicentrique en ouvert, randomisée, contrôlée contre comparateur
actif, en
deux bras, menée chez des patients présentant un cancer du sein
HER2-positif non résécable ou métastatique qui avaient reçu un
traitement antérieur
par trastuzumab et taxane pour la maladie métastatique ou qui avaient
présenté
une récidive de la maladie pendant un traitement adjuvant ou dans les
six mois
suivant la fin de celui-ci.
Des échantillons de tissu tumoral archivés étaient requis pour confirmer la positivité HER2, définie comme un score 3+ par immunohistochimie (IHC) ou un statut positif par hybridation in situ (HIS). Les patients ayant des antécédents de PID/pneumopathie inflammatoire ayant nécessité une corticothérapie ou présentant une PID/pneumopathie inflammatoire lors de la sélection, les patients présentant des métastases cérébrales non traitées et symptomatiques, les patients ayant des antécédents de cardiopathie cliniquement significative et les patients ayant reçu un traitement antérieur par un anticorps conjugué anti-HER2 pour la maladie métastatique étaient exclus de l'étude. Les patients ont été randomisés selon un rapport 1:1 pour recevoir Enhertu 5,4 mg/kg (N = 261) ou le trastuzumab emtansine 3,6 mg/kg (N = 263) en perfusion intraveineuse toutes les trois semaines. La randomisation était stratifiée en fonction du statut des récepteurs hormonaux, d'un traitement antérieur par le pertuzumab et de la présence d'un envahissement viscéral. Le traitement était administré jusqu'à progression de la maladie, décès, retrait du consentement ou survenue d'une toxicité inacceptable.
Le critère principal d'évaluation de l'efficacité était la survie sans progression (SSP) déterminée par revue centralisée indépendante en aveugle (RCIA) selon les Critères d'évaluation de la réponse dans les tumeurs solides (RECIST v1.1). La survie globale (SG) était le principal critère d'évaluation secondaire de l'efficacité. La SSP déterminée selon l'évaluation par les investigateurs, le taux de réponse objective (TRO) confirmée et la durée de la réponse (DR) étaient des critères secondaires.
Les caractéristiques démographiques et cliniques des patients à l'inclusion étaient équilibrées entre les bras de traitement. Chez les 524 patients randomisés, les caractéristiques démographiques et cliniques à l'inclusion étaient les suivantes : âge médian de 54 ans (plage : 20 à 83 ans), 65 ans et plus (20,2 %), femmes (99,6 %), asiatiques (59,9 %), blancs (27,3 %), noirs ou afro-américains (3,6 %), indice de performance ECOG (Eastern Cooperative Oncology Group) de 0 (62,8 %) ou de 1 (36,8 %), statut des récepteurs hormonaux (positif : 51,9 %), présence d'un envahissement viscéral (73,3 %), présence de métastases cérébrales lors de l'inclusion (15,6 %) et administration antérieure d'une ligne de traitement systémique pour la maladie métastatique (48,3 %). Le pourcentage de patients qui n'avaient pas reçu de traitement antérieur pour la maladie métastatique était de 9,5 %. Le pourcentage de patients préalablement traités par pertuzumab était de 61,1 %.
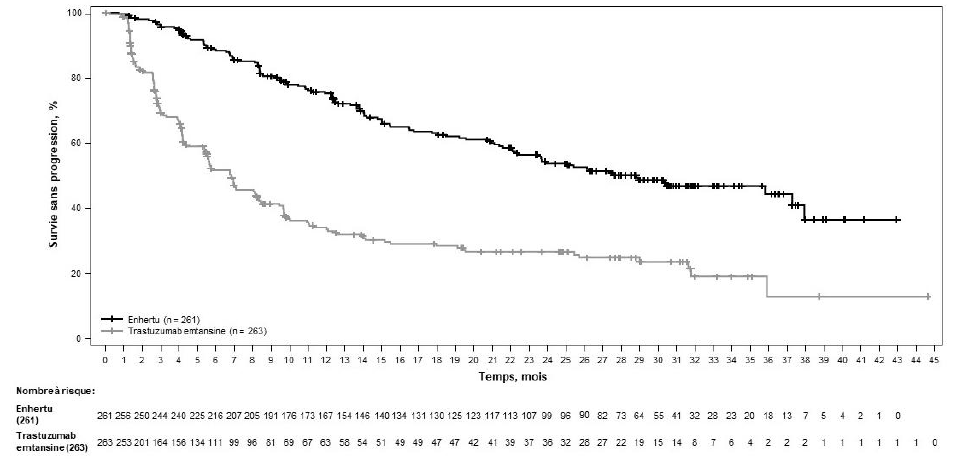
Au moment de l'analyse intermédiaire prédéfinie de la SSP basée sur 245 événements (73 % du nombre total d'événements prévu pour l'analyse finale), l'étude a montré une amélioration statistiquement significative de la SSP déterminée par RCIA chez les patients randomisés pour recevoir Enhertu par rapport aux patients recevant le trastuzumab emtansine. Les données de SSP déterminée par RCIA issues de l'analyse principale (date de gel des données : 21 mai 2021) et les résultats en termes de SG, de TRO et de DR actualisés issus des données disponibles à la date de gel des données du 25 juillet 2022 sont présentés dans le tableau 4.
Tableau 4 : Résultats d'efficacité dans l'étude DESTINY-Breast03
|
Critère d'efficacité |
Enhertu N = 261 |
Trastuzumab emtansine N = 263 |
|
Survie sans progression (SSP) déterminée par RCIAa |
||
|
Nombre d'événements (%) |
87 (33,3) |
158 (60,1) |
|
Médiane, mois (IC à 95 %) |
NA (18,5 ; NE) |
6,8 (5,6 ; 8,2) |
|
Hazard ratio (IC à 95 %) |
0,28 (0,22 ; 0,37) |
|
|
Valeur p |
p < 0,000001† |
|
|
Survie globale (SG)b |
||
|
Nombre d'événements (%) |
72 (27,6) |
97 (36,9) |
|
Médiane, mois (IC à 95 %) |
NA (40,5 ; NE) |
NA (34,0 ; NE) |
|
Hazard ratio (IC à 95 %) |
0,64 (0,47 ; 0,87) |
|
|
Valeur pc |
p = 0,0037 |
|
|
SSP déterminée par RCIA (données actualisées)b |
||
|
Nombre d'événements (%) |
117 (44,8) |
171 (65,0) |
|
Médiane, mois (IC à 95 %) |
28,8 (22,4 ; 37,9) |
6,8 (5,6 ; 8,2) |
|
Hazard ratio (IC à 95 %) |
0,33 (0,26 ; 0,43) |
|
|
Taux de réponse objective (TRO) confirmée déterminé par RCIAb |
||
|
n (%) |
205 (78,5) |
92 (35,0) |
|
IC à 95 % |
(73,1 ; 83,4) |
(29,2 ; 41,1) |
|
Réponse complète, n (%) |
55 (21,1) |
25 (9,5) |
|
Réponse partielle, n (%) |
150 (57,5) |
67 (25,5) |
|
Durée de la réponse déterminée par RCIAb |
||
|
Médiane, mois (IC à 95 %) |
36,6 (22,4 ; NE) |
23,8 (12,6 ; 34,7) |
IC = intervalle de confiance ; NA = non atteinte ; NE = non estimable.
† Présentée avec 6 décimales.
a Date de gel des données : 21 mai 2021.
b Date de gel des données pour une analyse intermédiaire de la SG prédéfinie : 25 juillet 2022.
c La valeur p a été déterminée selon un test du log-rank stratifié ; elle dépassait la limite d'efficacité de 0,013.
† Présentée avec 6 décimales.
a Date de gel des données : 21 mai 2021.
b Date de gel des données pour une analyse intermédiaire de la SG prédéfinie : 25 juillet 2022.
c La valeur p a été déterminée selon un test du log-rank stratifié ; elle dépassait la limite d'efficacité de 0,013.
Figure 1 : Courbe de Kaplan-Meier de la survie globale (date de gel des données : 25 juillet 2022)
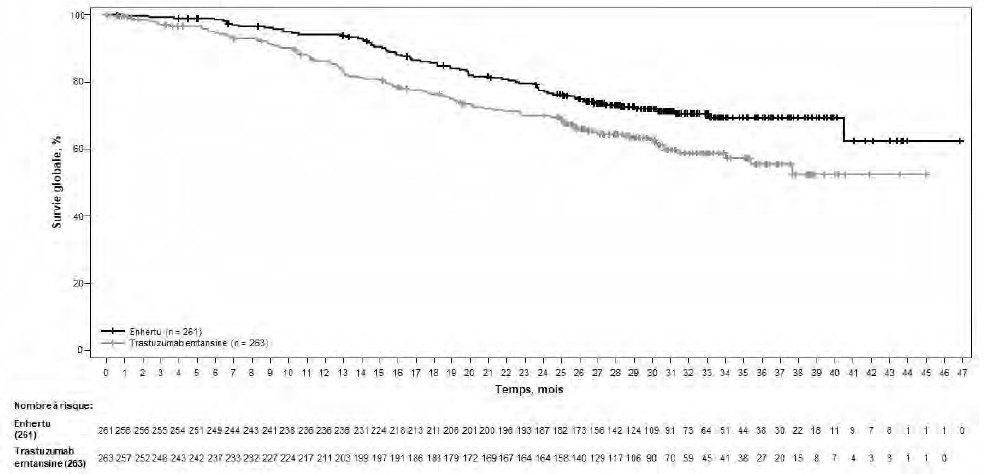
Figure 2 : Courbe de Kaplan-Meier de la survie sans progression déterminée par RCIA (date de gel des données : 25 juillet 2022)
Des résultats comparables en termes de SSP ont été observés dans les sous-groupes prédéfinis en fonction d'un traitement antérieur par le pertuzumab, du statut des récepteurs hormonaux et de la présence d'un envahissement viscéral.
DESTINY-Breast02 (NCT03523585)
L'efficacité
et la sécurité d'Enhertu ont été évaluées dans l'étude DESTINY-Breast02, une
étude de phase III multicentrique en ouvert, randomisée, contrôlée contre
comparateur actif, menée chez des patients présentant un cancer du sein
HER2-positif non résécable ou métastatique résistant ou réfractaire au
traitement antérieur par trastuzumab emtansine (T-DM1). Des échantillons de
tissu tumoral archivés étaient
requis pour confirmer la positivité HER2,
définie comme un score
3+ par IHC ou un statut positif par HIS. Les patients ayant des
antécédents de PID/pneumopathie inflammatoire ayant nécessité une
corticothérapie ou présentant une PID/pneumopathie inflammatoire lors de la
sélection, les patients présentant des métastases cérébrales non traitées et
symptomatiques et les patients ayant des antécédents
de cardiopathie cliniquement significative étaient exclus de l'étude. Les patients ont été randomisés
selon un rapport 2:1 pour recevoir Enhertu 5,4 mg/kg (n = 406) en perfusion
intraveineuse toutes les trois semaines ou le traitement choisi par le médecin
(n = 202, trastuzumab plus capécitabine ou lapatinib plus capécitabine).
La randomisation était
stratifiée en fonction du statut des récepteurs hormonaux, d'un traitement antérieur par le pertuzumab et
de la présence d'un envahissement viscéral. Le traitement était administré
jusqu'à progression de la maladie, décès, retrait du consentement ou survenue
d'une toxicité inacceptable.
Le critère principal d'évaluation de l'efficacité était la survie sans progression (SSP) déterminée par revue centralisée indépendante en aveugle (RCIA) selon les critères RECIST v1.1. La survie globale (SG) était le principal critère d'évaluation secondaire de l'efficacité. La SSP déterminée selon l'évaluation par les investigateurs, le taux de réponse objective (TRO) confirmée et la durée de la réponse (DR) étaient des critères secondaires.
Les caractéristiques démographiques et cliniques des patients à l'inclusion étaient comparables entre les bras de traitement. Chez les 608 patients randomisés, les caractéristiques étaient les suivantes : âge médian de 54 ans (plage : 22 à 88 ans), femmes (99,2 %), blancs (63,2 %), asiatiques (29,3 %), noirs ou afro-américains (2,8 %), indice de performance ECOG (Eastern Cooperative Oncology Group) de 0 (57,4 %) ou de 1 (42,4%), statut des récepteurs hormonaux positif (58,6 %), présence d'un envahissement viscéral (78,3 %), présence de métastases cérébrales lors de l'inclusion (18,1 %) et administration antérieure d'une ligne de traitement systémique pour la maladie métastatique (4,9 %).
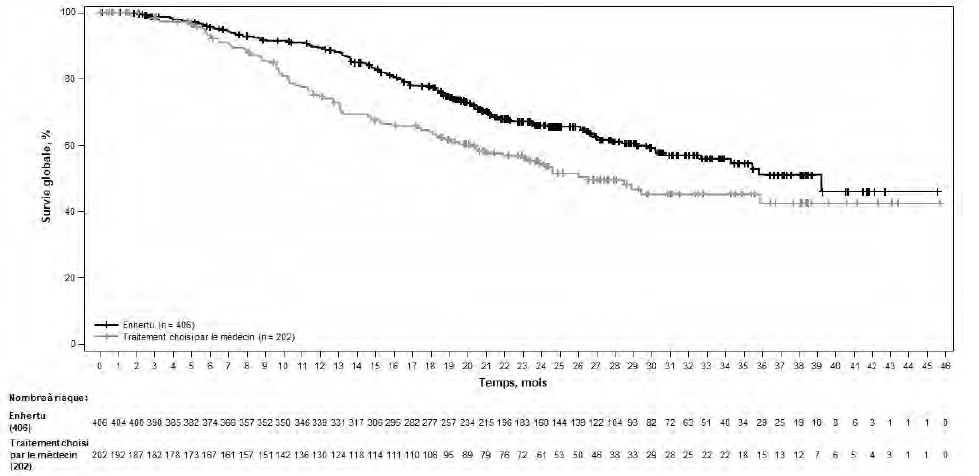
Les résultats d'efficacité sont présentés dans le tableau 5 et les figures 3 et 4.
Tableau 5 : Résultats d'efficacité dans l'étude DESTINY-Breast02
|
Critère d'efficacité |
Enhertu N = 406 |
Traitement choisi
par le médecin N = 202 |
|
SSP déterminée par RCIA |
||
|
Nombre d'événements (%) |
200 (49,3) |
125 (61,9) |
|
Médiane, mois (IC à 95 %) |
17,8 (14,3 ; 20,8) |
6,9 (5,5 ; 8,4) |
|
Hazard ratio (IC à 95 %) |
0,36 (0,28 ; 0,45) |
|
|
Valeur p |
p < 0,000001† |
|
|
Survie globale (SG) |
||
|
Nombre d'événements (%) |
143 (35,2) |
86 (42,6) |
|
Médiane, mois (IC à 95 %) |
39,2 (32,7 ; NE) |
26,5 (21,0 ; NE) |
|
Hazard ratio (IC à 95 %) |
0,66 (0,50 ; 0,86) |
|
|
Valeur pa |
p = 0,0021 |
|
|
SSP déterminée selon l'évaluation par les investigateurs |
||
|
Nombre d'événements (%) |
206 (50,7) |
152 (75,2) |
|
Médiane, mois (IC à 95 %) |
16,7 (14,3 ; 19,6) |
5,5 (4,4 ; 7,0) |
|
Hazard ratio (IC à 95 %) |
0,28 (0,23 ; 0,35) |
|
|
Taux de réponse objective (TRO) confirmée déterminé par RCIA |
||
|
n (%) |
283 (69,7) |
59 (29,2) |
|
IC à 95 % |
(65,0 ; 74,1) |
(23,0 ; 36,0) |
|
Réponse complète, n (%) |
57 (14,0) |
10 (5,0) |
|
Réponse partielle, n (%) |
226 (55,7) |
49 (24,3) |
|
Durée de la réponse déterminée par RCIA |
||
|
Médiane, mois (IC à 95 %) |
19,6 (15,9 ; NE) |
8,3 (5,8 ; 9,5) |
IC, intervalle de confiance ; NE, non estimable.
† Présentée avec 6 décimales.
a La valeur p a été déterminée selon un test du log-rank stratifié ; elle dépassait la limite d'efficacité de 0,004.
† Présentée avec 6 décimales.
a La valeur p a été déterminée selon un test du log-rank stratifié ; elle dépassait la limite d'efficacité de 0,004.
Figure 3 : Courbe de Kaplan-Meier de la survie sans progression déterminée par RCIA

Figure 4 : Courbe de Kaplan-Meier de la survie globale
DESTINY-Breast01 (NCT03248492)
L'efficacité
et la sécurité d'Enhertu ont été évaluées dans l'étude DESTINY-Breast01, une
étude de phase II multicentrique en ouvert en un seul bras, menée chez des
patientes présentant un cancer du sein HER2-positif non résécable et/ou
métastatique qui avaient reçu préalablement au moins deux lignes de traitement
comportant un agent anti-HER2, dont le trastuzumab emtansine (100 %), le
trastuzumab (100 %) et le pertuzumab (65,8 %). Des échantillons de tissu
tumoral archivés étaient requis pour confirmer la positivité HER2, définie
comme un score 3+ par immunohistochimie (IHC) ou un statut positif par
hybridation in situ (HIS). Les patientes ayant des antécédents de PID ou
présentant une PID lors de la sélection, les patientes présentant des
métastases cérébrales non traitées ou symptomatiques et les patientes ayant des
antécédents de cardiopathie cliniquement significative étaient exclues de
l'étude. Les patientes incluses présentaient au moins une lésion mesurable
selon les critères RECIST v1.1. Enhertu était administré en perfusion
intraveineuse à la dose de 5,4 mg/kg toutes les trois semaines jusqu'à
progression de la maladie, décès, retrait du consentement ou survenue
d'une toxicité inacceptable. Le critère d'évaluation principal de l'efficacité
était le taux de réponse objective (TRO) confirmée selon les critères RECIST
v1.1 dans la population en intention de traiter, évaluée par revue centralisée indépendante (RCI). Le critère
d'efficacité secondaire était la
durée de la réponse (DR).
Les caractéristiques démographiques et cliniques à l'inclusion des 184 patientes incluses dans l'étude DESTINY-Breast01 étaient : âge médian de 55 ans (plage : 28 à 96 ans), 65 ans et plus (23,9 %), femmes (100 %), blanches (54,9 %), asiatiques (38,0 %), noires ou afro-américaines (2,2 %), indice de performance ECOG (Eastern Cooperative Oncology Group) de 0 (55,4 %) ou de 1 (44,0 %), statut des récepteurs hormonaux (positif : 52,7 %), présence d'un envahissement viscéral (91,8 %), métastases cérébrales préalablement traitées et stables (13,0 %), nombre médian de traitements antérieurs de la maladie métastatique : 5 (plage : 2 à 17), somme des diamètres des lésions cibles (< 5 cm : 42,4 %,≥ 5 cm : 50,0 %).
Une analyse préliminaire (durée médiane de suivi de 11,1 mois [plage : 0,7 à 19,9 mois]) a montré un taux de réponse objective confirmée de 60,9 % (IC à 95 % : 53,4 ; 68,0), 6,0 % de patientes ayant obtenu une réponse complète et 54,9 % une réponse partielle ; 36,4 % des patientes présentaient une maladie stable, 1,6 % une progression de la maladie et 1,1 % n'étaient pas évaluables. La durée médiane de réponse à cette date était de 14,8 mois (IC à 95 % : 13,8 ; 16,9), avec une durée de réponse ≥ 6 mois chez 81,3 % des patientes ayant obtenu une réponse (IC à 95 % : 71,9 ; 87,8). Les résultats d'efficacité issues d'une analyse actualisée au moment du gel des données avec une durée médiane de suivi de 20,5 mois (plage : 0,7 à 31,4 mois) sont présentés dans le tableau 6.
Tableau 6 : Résultats d'efficacité dans l'étude DESTINY-Breast01 (population en intention de traiter)
|
|
DESTINY-Breast01 N = 184 |
|
Taux de réponse objective confirmée (IC à 95 %)*† |
61,4 % (54,0 ; 68,5) |
|
Réponse complète (RC) |
6,5 % |
|
Réponse partielle (RP) |
54,9 % |
|
Durée de la réponse‡ |
|
|
Médiane, mois (IC à 95 %) |
20,8 (15,0 ; NA) |
|
% de patientes avec durée de la réponse ≥ 6 mois (IC à 95 %)§ |
81,5 % (72,2 ; 88,0) |
IC à 95 % du TRO calculé
selon la méthode
de Clopper-Pearson.
IC = intervalle de confiance.
IC à 95 % calculés selon la méthode de Brookmeyer-Crowley.
* Les réponses confirmées (par revue centralisée indépendante en aveugle) étaient définies comme une RC ou RP enregistrée, confirmée par de nouveaux examens d'imagerie réalisés au moins 4 semaines après la visite au cours de laquelle la réponse avait été observée pour la première fois.
† Sur les 184 patientes, 35,9 % présentaient une maladie stable, 1,6 % une progression de la maladie et 1,1 % n'étaient pas évaluables.
‡ Inclut 73 patientes dont les données ont été censurées.
§ Selon une estimation de Kaplan-Meier.
NA = non atteinte.
IC = intervalle de confiance.
IC à 95 % calculés selon la méthode de Brookmeyer-Crowley.
* Les réponses confirmées (par revue centralisée indépendante en aveugle) étaient définies comme une RC ou RP enregistrée, confirmée par de nouveaux examens d'imagerie réalisés au moins 4 semaines après la visite au cours de laquelle la réponse avait été observée pour la première fois.
† Sur les 184 patientes, 35,9 % présentaient une maladie stable, 1,6 % une progression de la maladie et 1,1 % n'étaient pas évaluables.
‡ Inclut 73 patientes dont les données ont été censurées.
§ Selon une estimation de Kaplan-Meier.
NA = non atteinte.
Une activité antitumorale uniforme a été observée dans les sous-groupes prédéfinis en fonction d'un traitement antérieur par le pertuzumab et du statut des récepteurs hormonaux.
Cancer du sein HER2-faible et HER2-ultrafaible
DESTINY-Breast06 (NCT04494425)
L'efficacité
et la sécurité d'Enhertu ont été évaluées dans l'étude DESTINY-Breast06, une
étude de phase III multicentrique randomisée en ouvert, menée chez 866 patients adultes
présentant un cancer du sein RH+ avancé ou métastatique
HER2-faible (score IHC 1+ ou IHC 2+/HIS-) ou HER2- ultrafaible, déterminée par un laboratoire central par
test PATHWAY/VENTANA anti-HER2/neu (4B5). Le statut HER2-ultrafaible (IHC 0
avec marquage membranaire, établi par un score IHC > 0 < 1+ dans l'étude)
est défini par un marquage
membranaire faible et incomplet
d'HER2, observé
dans 10 % ou moins des cellules tumorales. Les patients étaient éligibles s'ils avaient présenté une progression de la maladie
sous (a) au moins deux lignes d'hormonothérapie pour la maladie métastatique ou
(b) une ligne d'hormonothérapie pour la maladie métastatique et avaient
présenté une progression de la maladie dans les 24 mois suivant le début de
l'hormonothérapie adjuvante ou dans les 6 mois suivant le début de la première
ligne d'hormonothérapie en association avec un inhibiteur de CDK4/6 en situation métastatique. Les
patients ayant reçu préalablement une chimiothérapie en situation néoadjuvante
ou adjuvante étaient éligibles si le délai sans récidive était supérieur à 12
mois. Les patients ayant reçu préalablement une chimiothérapie pour la maladie avancée
ou métastatique, les patients ayant des antécédents de
PID/pneumopathie inflammatoire ayant nécessité
une corticothérapie ou présentant une PID/ pneumopathie inflammatoire
lors de la sélection, les patients présentant une maladie cardiovasculaire non
contrôlée ou significative, des métastases cérébrales non traitées et
symptomatiques ou ayant un indice de performance ECOG > 1 étaient exclus de
l'étude.
Les patients ont été randomisés selon un rapport 1:1 pour recevoir Enhertu 5,4 mg/kg (N = 436) en perfusion intraveineuse toutes les trois semaines ou la monochimiothérapie choisie par le médecin (N = 430, capécitabine 60 %, nab-paclitaxel 24 %, ou paclitaxel 16 %). La randomisation était stratifiée en fonction du traitement antérieur par un inhibiteur de CDK4/6 (oui ou non), du traitement antérieur par taxane en situation non métastatique (oui ou non) et du statut HER2 déterminé par IHCsur des échantillons de tissu tumoral (IHC 2+/HIS-, IHC 1+, IHC > 0 < 1+). Le traitement par Enhertu était administré jusqu'à progression de la maladie, décès, retrait du consentement ou survenue d'une toxicité inacceptable.
Le critère d'évaluation principal de l'efficacité était la SSP chez les patients présentant un cancer du sein HER2-faible, évaluée par RCIA selon les critères RECIST v1.1. Les critères secondaires d'efficacité hiérarchisés étaient la SSP évaluée par RCIA selon les critères RECIST v1.1 dans la population totale (HER2-faible et HER2-ultrafaible), la SG chez les patients HER2-faible et la SG dans la population totale. Le TRO et la DR étaient des critères d'évaluation secondaires.
Dans la population totale, les caractéristiques démographiques et cliniques des patients à l'inclusion étaient comparables entre les bras de traitement. Chez les 866 patients randomisés, l'âge médian était de 57 ans (plage : 28 à 87 ans) ; 31 % étaient âgés de 65 ans ou plus ; 99,9 % étaient des femmes ; 53 % étaient blancs, 35 % étaient asiatiques et 1 % étaient noirs ou afro-américains. Les patients avaient un indice de performance ECOG de 0 (59 %) ou 1 (39 %) lors de l'inclusion ; 18 % avaient un score IHC > 0 < 1+, 55 % un score IHC 1+, 27 % un score IHC 2+/HIS- ; 67 % présentaient des métastases hépatiques, 32 % des métastases pulmonaires, 8 % des métastases cérébrales et 3 % des métastases osseuses uniquement. Les patients avaient reçu un nombre médian de deux lignes d'hormonothérapie en situation métastatique (plage : 1 à 5), 17 % d'entre eux en ayant reçu une et 68 % en ayant reçu deux. Quatre-vingt-neuf pour cent des patients avaient reçu préalablement une hormonothérapie en association avec un inhibiteur de CDK4/6 en situation métastatique, 47 % avaient reçu préalablement une anthracycline et 41 % avaient déjà reçu un taxane en situation non métastatique.
Les résultats d'efficacité sont présentés dans le tableau 7 et les figures 5 et 6.
Tableau 7: Résultats d'efficacité dans l'étude DESTINY-Breast06
|
Critère d'efficacité |
HER2-faible |
Population totale (HER2-faible et HER2-ultrafaible) |
||
|
Enhertu (N = 359) |
Chimiothérapie (N = 354) |
Enhertu (N = 436) |
Chimiothérapie (N = 430) |
|
|
Survie sans progression déterminée par RCIA |
||||
|
Nombre d'événements (%) |
225 (62,7) |
232 (65,5) |
269 (61,7) |
271 (63,0) |
|
Médiane, mois (IC à 95 %) |
13,2 (11,4 ; 15.2) |
8,1 (7,0 ; 9,0) |
13,2 (12.0 ; 15,2) |
8,1 (7.0 ; 9,0) |
|
Hazard ratio (IC à 95 %) |
0,62 (0,52 ; 0,75) |
0.64 (0,54 ; 0,76) |
||
|
Valeur p |
< 0,0001 |
< 0,0001 |
||
|
Survie globale* |
||||
|
Nombre d'événements (%) |
136 (37,9) |
146 (41,2) |
161 (36,9) |
174 (40,5) |
|
Médiane, mois (IC à 95 %) |
28,9 (25,7 ; 33,7) |
27,1 (23,5 ; 29,9) |
28,9 (26,4 ; 32,7) |
27,4 (23,9 ; 29,9) |
|
Hazard ratio (IC à 95 %) |
0,83 (0,66 ; 1,05) |
0,81 (0,66 ; 1,01) |
||
|
Taux de réponse objective (TRO) confirmée déterminé par RCIA† |
||||
|
n (%) |
203 (56,5) |
114 (32,2) |
250 (57,3) |
134 (31,2) |
|
IC à 95 % |
51,2 ; 61,7 |
27,4 ; 37,3 |
52,5 ; 62,0 |
26,8 ; 35,8 |
|
Durée de la réponse déterminée par RCIA† |
||||
|
Médiane, mois (IC à 95 %) |
14,1 (11,8 ; 15,9) |
8,6 (6,7 ; 11,3) |
14,3 (12,5 ; 15,9) |
8,6 (6,9 ; 11,5) |
Date de gel des données
: 18 mars 2024.
IC = intervalle de confiance.
* Première analyse intermédiaire planifiée.
† Les résultats n'ont pas fait l'objet d'un contrôle du risque alpha et doivent être interprétés comme des statistiques descriptives.
IC = intervalle de confiance.
* Première analyse intermédiaire planifiée.
† Les résultats n'ont pas fait l'objet d'un contrôle du risque alpha et doivent être interprétés comme des statistiques descriptives.
Un bénéfice homogène en termes de SSP a été observé dans les sous-groupes pré-spécifiés, y compris en fonction de l'expression d'HER2 (IHC > 0 < 1+, IHC 1+, IHC 2+/HIS-), du traitement antérieur par inhibiteur de CDK4/6 (oui ou non), de l'utilisation antérieure d'un taxane en situation non métastatique (oui ou non) et du nombre de lignes antérieures d'hormonothérapie en situation métastatique.
Dans le sous-groupe HER2-ultrafaible (n = 152), la SSP médiane était de 13,2 mois (IC à 95 % : 9,8 ; 17,3) chez les patients randomisés pour recevoir Enhertu (N = 76) et de 8,3 mois (IC à 95 % : 5,8 ; 15,2) chez les patients randomisés pour recevoir la chimiothérapie, avec un hazard ratio de 0,78 (IC à 95 % : 0,50 ; 1,21). La SG médiane était de 29,5 mois (IC à 95 % : 27,9 ; non estimable) chez les patients randomisés pour recevoir Enhertu et de 27,4 mois (IC à 95 % : 19,4 ; non estimable) chez les patients randomisés pour recevoir la chimiothérapie, avec un hazard ratio de 0,75 (IC à 95 % : 0,43 ; 1,29). Le taux de réponse objective confirmée était de 61,8 % (IC à 95 % : 50,0 ; 72,8) et de 26,3 % (IC à 95 % : 16,9 ; 37,7) chez les patients randomisés pour recevoir Enhertu et la chimiothérapie, respectivement. La durée médiane de réponse était de 14,3 mois (IC à 95 % : 9,2 ; 20,7) et de14,1 mois (IC à 95 % : 5,9 ; non estimable) chez les patients randomisés pour recevoir Enhertu et la chimiothérapie, respectivement.
Figure 5 : Courbe de Kaplan-Meier de la survie sans progression (population totale)
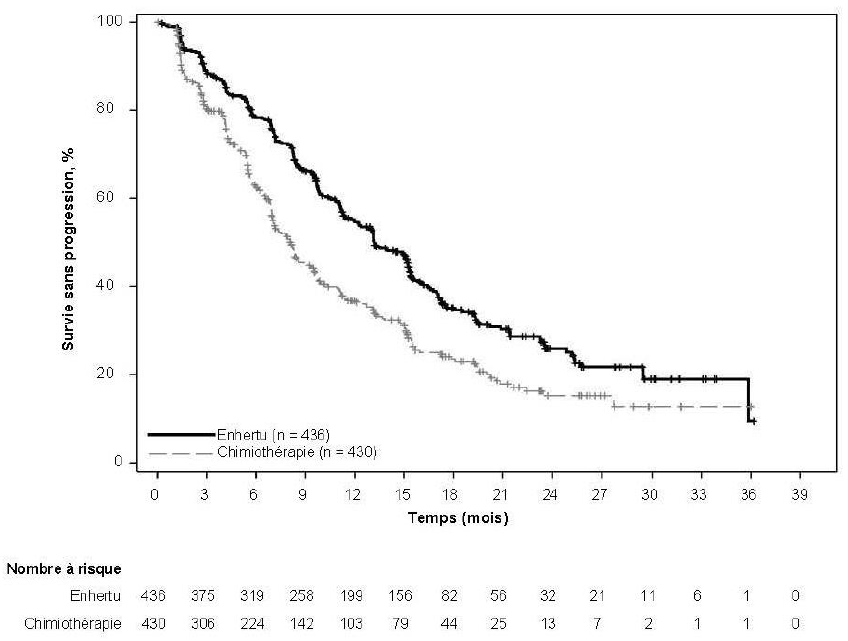
Figure 6 : Courbe de Kaplan-Meier de la survie globale (population totale)
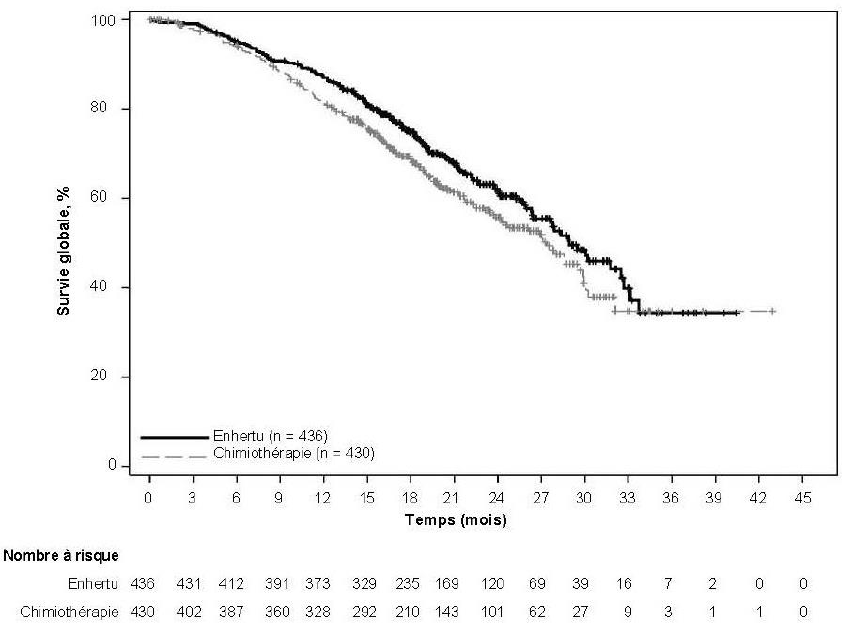
DESTINY-Breast04 (NCT03734029)
L'efficacité
et la sécurité d'Enhertu ont été évaluées dans l'étude DESTINY-Breast04, une
étude de phase III multicentrique randomisée en ouvert,
menée chez 557 patients adultes
présentant un cancer du sein HER2-faible non résécable ou métastatique. L'étude comportait deux cohortes : 494
patients avec statut positif pour les récepteurs hormonaux (RH+) et 63 patients
avec statut négatif (RH-).
L'expression
faible d'HER2 était définie comme un score IHC 1+ (défini par un
marquage
membranaire partiel faible dans plus de 10 % des cellules tumorales) ou
IHC
2+/HIS-, déterminé par un laboratoire central à l'aide du test
PATHWAY/VENTANA
anti-HER2/neu (4B5). Les patients devaient avoir reçu une
chimiothérapie pour la maladie métastatique ou avoir présenté une
récidive de la maladie pendant la
chimiothérapie adjuvante ou au cours des six mois suivant la fin de la
chimiothérapie adjuvante. Conformément aux critères d'inclusion, les
patients
ayant le statut RH+ devaient avoir reçu au moins une hormonothérapie et
être
inéligibles à une autre hormonothérapie au moment de la randomisation.
Les
patients ont été randomisés selon un rapport 2:1 pour recevoir Enhertu
5,4 mg/kg
(N = 373) en perfusion intraveineuse toutes les trois semaines ou la
chimiothérapie
choisie par le médecin (N = 184, éribuline 51,1 %, capécitabine 20,1 %,
gemcitabine 10,3 %, nab- paclitaxel 10,3 % ou paclitaxel 8,2 %). La
randomisation était stratifiée en fonction du statut HER2 déterminé par
IHC sur
des échantillons de tissu tumoral (IHC 1+ ou IHC 2+/HIS-), du nombre de
lignes
de chimiothérapie antérieures en situation métastatique (1 ou 2) et du
statut
des RH/du traitement antérieur par un inhibiteur de CDK4/6 (RH+ avec
traitement
anti-CDK4/6 antérieur, RH+ sans traitement anti-CDK4/6 antérieur ou
RH-). Le
traitement était administré jusqu'à progression de la maladie, décès,
retrait
du consentement ou survenue d'une toxicité inacceptable. Les patients
ayant des antécédents de PID/pneumopathie inflammatoire ayant nécessité
une corticothérapie ou présentant
une PID/pneumopathie inflammatoire lors de la sélection et les patients
présentant une cardiopathie cliniquement significative étaient exclus
de
l'étude. Les patients présentant des métastases cérébrales non traitées
ou
symptomatiques ou ayant un indice de performance ECOG > 1 étaient
également exclus.
Le critère d'évaluation principal de l'efficacité était la survie sans progression (SSP) chez les patients présentant un cancer du sein avec statut RH+, évaluée par RCIA selon les critères RECIST v1.1. Les principaux critères d'évaluation secondaires de l'efficacité étaient la SSP évaluée par RCIA selon les critères RECIST v1.1 dans la population totale (ensemble des patients avec statut RH+ ou RH- randomisés), la survie globale (SG) chez les patients avec statut RH+ et la SG dans la population totale. Le TRO, la DR et les scores de l'échelle d'auto-évaluation par les patients (PRO - patient- reported outcomes) étaient des critères d'évaluation secondaires.
Les caractéristiques démographiques et cliniques des patients à l'inclusion étaient comparables entre les bras de traitement. Chez les 557 patients randomisés, l'âge médian était de 57 ans (plage : 28 à 81 ans), 23,5 % étaient âgés de 65 ans et plus, 99,6 % étaient des femmes et 0,4 % étaient des hommes, 47,9 % étaient blancs, 40,0 % étaient asiatiques et 1,8 % étaient noirs ou afro-américains. Les patients avaient un indice de performance ECOG de 0 (54,8 %) ou 1 (45,2 %) lors de l'inclusion, 56,7 % avaient un score IHC 1+, 42,4 % un score IHC 2+/HIS-, 88,7 % avaient le statut RH+ et 11,3 % le statut RH-, 69,8 % présentaient des métastases hépatiques, 32,9 % des métastases pulmonaires et 5,7 % des métastases cérébrales. Les pourcentages de patients ayant reçu un traitement antérieur par anthracycline étaient de 46,3 % en situation (néo)adjuvante et de 19,4 % en situation de maladie localement avancée et/ou métastatique. En situation métastatique, les patients avaient reçu un nombre médian de trois lignes de traitement systémique antérieures (plage : 1 à 9), 57,6 % des patients ayant reçu un protocole de chimiothérapie antérieur et 40,9 % des patients en ayant reçu deux et 3,9 % des patients présentaient une progression rapide de la maladie (progression en situation néo/adjuvante). Chez les patients ayant le statut RH+, le nombre médian de lignes d'hormonothérapie antérieures étaient de 2 (plage : 0 à 9) et 70 % des patients avaient reçu un traitement antérieur par un inhibiteur de CDK4/6.
Les résultats d'efficacité sont présentés dans le tableau 8 et les figures 7 et 8.
Tableau 8 : Résultats d'efficacité dans l'étude DESTINY-Breast04
|
Critère d'efficacité |
Cohorte RH+ |
Population
totale (cohortes RH+ et RH-) |
||
|
Enhertu (N = 331) |
Chimiothérapie (N = 163) |
Enhertu (N = 373) |
Chimiothérapie (N = 184) |
|
|
Survie globale |
||||
|
Nombre d'événements (%) |
126 (38,1) |
73 (44,8) |
149 (39,9) |
90 (48,9) |
|
Médiane, mois (IC à 95 %) |
23,9 (20,8 ; 24,8) |
17,5 (15,2 ; 22,4) |
23,4 (20,0 ; 24,8) |
16,8 (14,5 ; 20,0) |
|
Hazard ratio (IC à 95 %) |
0,64 (0,48 ; 0,86) |
0,64 (0,49 ; 0,84) |
||
|
Valeur p |
0,0028 |
0,001 |
||
|
Survie sans progression déterminée par RCIA |
||||
|
Nombre d'événements (%) |
211 (63,7) |
110 (67,5) |
243 (65,1) |
127 (69,0) |
|
Médiane, mois (IC à 95 %) |
10,1 (9,5 ; 11,5) |
5,4 (4,4 ; 7,1) |
9,9 (9,0 ; 11,3) |
5,1 (4,2 ; 6,8) |
|
Hazard ratio (IC à 95 %) |
0,51 (0,40 ; 0,64) |
0,50 (0,40 ; 0,63) |
||
|
Valeur p |
< 0,0001 |
< 0,0001 |
||
|
Taux de réponse objective (TRO) confirmée déterminé par RCIA* |
||||
|
n (%) |
175 (52,6) |
27 (16,3) |
195 (52,3) |
30 (16,3) |
|
IC à 95 % |
47,0 ; 58,0 |
11,0 ; 22,8 |
47,1 ; 57,4 |
11,3 ; 22,5 |
|
Réponse complète, n (%) |
12 (3,6) |
1 (0,6) |
13 (3,5) |
2 (1,1) |
|
Réponse partielle, n (%) |
164 (49,2) |
26 (15,7) |
183 (49,1) |
28 (15,2) |
|
Durée de la réponse déterminée par RCIA* |
||||
|
Médiane, mois (IC à 95 %) |
10,7 (8,5 ; 13,7) |
6,8 (6,5 ; 9,9) |
10,7 (8,5 ; 13,2) |
6,8 (6,0 ; 9,9) |
IC = intervalle de confiance.
* Sur la base des données issues des cahiers d'observation électroniques pour la cohorte RH+ : N = 333 dans le bras Enhertu et N = 166 dans le bras chimiothérapie.
* Sur la base des données issues des cahiers d'observation électroniques pour la cohorte RH+ : N = 333 dans le bras Enhertu et N = 166 dans le bras chimiothérapie.
Un bénéfice homogène en termes de SG et de SSP a été observé dans les sous-groupes prédéfinis en fonction du statut des RH, du traitement antérieur ou non par un inhibiteur de CDK4/6, du nombre de chimiothérapies antérieures et du statut IHC 1+ ou IHC 2+/HIS-. Dans le sous-groupe RH-, la SG médiane était de 18,2 mois (IC à 95 % : 13,6 ; non estimable) chez les patients randomisés pour recevoir Enhertu contre 8,3 mois (IC à 95 % : 5,6 ; 20,6) chez les patients randomisés pour recevoir la chimiothérapie, avec un hazard ratio de 0,48 (IC à 95 % : 0,24 ; 0,95). La SSP médiane était de 8,5 mois (IC à 95 % : 4,3 ; 11,7) chez les patients randomisés pour recevoir Enhertu et de 2,9 mois (IC à 95 % : 1,4 ; 5,1) chez les patients randomisés pour recevoir la chimiothérapie, avec un hazard ratio de 0,46 (IC à 95 % : 0,24 ; 0,89).
Dans une analyse descriptive actualisée avec une durée médiane de suivi de 32 mois, les améliorations de la SG concordaient avec les résultats de l'analyse principale. Le hazard ratio dans la populationtotale était de 0,69 (IC à 95 % : 0,55 ; 0,86), avec une SG médiane de 22,9 mois (IC à 95 % : 21,2 ; 24,5) dans le bras recevant Enhertu contre 16,8 mois (IC à 95 % : 14,1 ; 19,5) dans le bras recevant la chimiothérapie. La courbe de Kaplan-Meier de la SG selon l'analyse actualisée est présentée dans la figure 7.
Figure 7 : Courbe de Kaplan-Meier de la survie globale (population totale) (analyse actualisée)
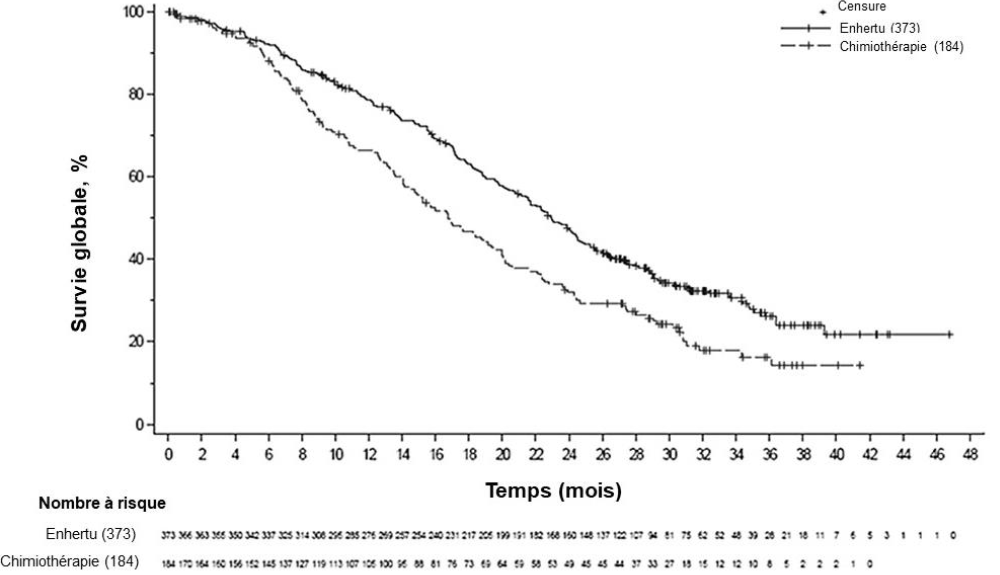
Figure 8 : Courbe de Kaplan-Meier de la survie sans progression déterminée par RCIA (population totale)
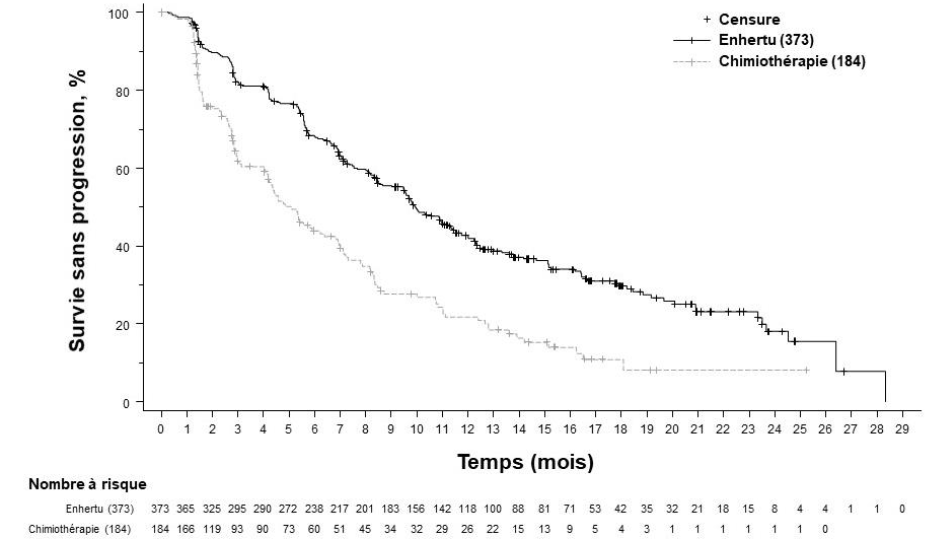
CBNPC
DESTINY-Lung02 (NCT04644237)
L'efficacité et la sécurité d'Enhertu ont été évaluées
dans l'étude DESTINY-Lung02, une étude dephase II randomisée visant à évaluer
deux doses en double aveugle (patients
et investigateurs). L'étude a été menée chez des patients
adultes présentant un CBNPC métastatique avec mutation d'HER2 qui avaient reçu
au moins une ligne de traitement comportant une chimiothérapie à base de
platine. La présence d'une mutation activatrice d'HER2 (ERBB2) était déterminée
prospectivement dans les échantillons de tissu tumoral par le laboratoire local
à l'aide d'une méthode validée telle que séquençage à haut débit, test PCR
(réaction en chaîne par polymérase) ou spectrométrie
de masse. Les patients ont été randomisés selon un rapport 2:1 pour recevoir
Enhertu 5,4 mg/kg ou 6,4 mg/kg toutes les trois semaines. La randomisation
était stratifiée en fonction d'un traitement antérieur par un inhibiteur de
PD-1 (programmed cell death receptor-1) et/ou de PD-L1 (programmed
cell death ligand 1) (oui versus
non). Le traitement était administré jusqu'à progression de la maladie,
décès, retrait du consentement ou survenue
d'une toxicité inacceptable. Les patients ayant des antécédents de
PID/pneumopathie inflammatoire ayant nécessité une corticothérapie ou
présentant une PID/pneumopathie inflammatoire lors de la sélection et les
patients présentant une cardiopathie cliniquement significative étaient exclus
de l'étude. Les patients présentant des métastases cérébrales non traitées et
symptomatiques ou ayant un indice de performance ECOG > 1 étaient également exclus.
Le critère d'évaluation principal de l'efficacité était le TRO confirmée, déterminée par RCIA selon les critères RECIST v1.1. Le critère d'efficacité secondaire était la durée de la réponse (DR).
Les caractéristiques démographiques et cliniques à l'inclusion des 102 patients inclus dans le bras recevant la dose de 5,4 mg/kg étaient : âge médian de 59,4 ans (plage : 31 à 84 ans), femmes (63,7 %), asiatiques (63,7 %), blancs (22,5 %) ou autre groupe ethnique (13,7 %), indice de performance ECOG de 0 (28,4 %) ou de 1 (71,6 %), présence d'une mutation dans le domaine kinase d'ERBB2 (97,1 %), présence d'une mutation dans le domaine extracellulaire (2,9 %), présence d'une mutation d'HER2 dans l'exon 19 ou l'exon 20 (96,1 %), présence de métastases cérébrales stables (34,3 %), anciens fumeurs (46,1 %), fumeurs actuels (0 %), antécédents de résection pulmonaire (21,6%). Pour la maladie métastatique, 32,4 % avaient reçu plus de deux lignes de traitement systémique antérieures, 100 % avaient reçu une chimiothérapie à base de platine, 73,5 % un inhibiteur de PD-1/PD-L1 et 50,0 % un traitement antérieur consistant en une chimiothérapie à base de platine et un inhibiteur de PD-1/PD-L1 en association.
Les résultats d'efficacité sont présentés dans le tableau 9. La durée médiane de suivi était de 11,5 mois (date de gel des données : 23 décembre 2022).
Tableau 9 : Résultats d'efficacité dans l'étude DESTINY-Lung02
|
Critère d'efficacité |
DESTINY-Lung02 5,4 mg/kg N = 102 |
|
Taux de réponse objective (TRO) confirmée déterminé par RCIA |
|
|
n (%) |
50 (49,0) |
|
(IC à 95 %)* |
(39,0 ; 59,1) |
|
Réponse complète (RC), n (%) |
1 (1,0) |
|
Réponse partielle (RP), n (%) |
49 (48,0) |
|
Durée de la réponse |
|
|
Médiane, mois (IC à 95 %)† |
16,8 (6,4 ; NE) |
* IC à 95 % calculé selon la méthode
de Clopper-Pearson.
IC, intervalle de confiance ; NE, non estimable.
† IC à 95 % calculé selon la méthode de Brookmeyer-Crowley.
IC, intervalle de confiance ; NE, non estimable.
† IC à 95 % calculé selon la méthode de Brookmeyer-Crowley.
Cancer de l'estomac
DESTINY-Gastric02 (NCT04014075)
L'efficacité
et la sécurité d'Enhertu ont été évaluées dans l'étude DESTINY-Gastric02, une
étude de phase II multicentrique en ouvert en un seul bras menée en Europe et aux États-Unis. Dans l'étude ont été inclus
des patients présentant un adénocarcinome gastrique ou de la jonction
œsogastrique (JOG) HER2 positif localement avancé ou métastatique qui avait
progressé sous une ligne de traitement comportant le trastuzumab. Les échantillons
tumoraux devaient présenter une positivité HER2 confirmée de manière
centralisée, définie comme un score IHC 3+ ou un score IHC 2+/HIS+. Les
patients ayant des antécédents de PID/pneumopathie inflammatoire ayant nécessité une corticothérapie
ou présentant une PID/pneumopathie inflammatoire lors de la sélection, les
patients ayant des antécédents de cardiopathie cliniquement significative et
les patients présentant des métastases cérébrales évolutives étaient exclus de l'étude.
Enhertu était administré en
perfusion intraveineuse à la dose de
6,4 mg/kg toutes les trois semaines jusqu'à progression de la maladie, décès,
retrait du consentement ou survenue d'une toxicité inacceptable. Le critère
d'évaluation principal de l'efficacité était le taux de réponse objective (TRO)
confirmée, évaluée par RCI selon les critères RECIST v1.1.
La DR et la SG étaient
des critères d'évaluation secondaires.
Chez les 79 patients inclus dans l'étude DESTINY-Gastric02, les caractéristiques démographiques et cliniques à l'inclusion étaient : l'âge médian était de 61 ans (plage : 20 à 78 ans), 72 % étaient des hommes, 87 % des patients étaient blancs, 5,0 % étaient asiatiques et 1,0 % étaient noirs ou afro- américains. Les patients avaient un indice de performance ECOG de 0 (37 %) ou 1 (63 %), 34 % présentaient un adénocarcinome de l'estomac et 66 % un adénocarcinome de la JOG, 86 % avaient un score IHC 3+ et 13 % un score IHC 2+/HIS+ et 63 % présentaient des métastases hépatiques.
Les résultats d'efficacité en termes de TRO, de DR et de SG sont présentés dans le tableau 10.
Tableau 10 : Résultats d'efficacité dans l'étude DESTINY-Gastric02 (population complète d'analyse, FAS*)
|
Critère d'efficacité |
DESTINY-Gastric02 N = 79 |
|
Date de gel des données : 8 novembre 2021 |
|
|
Taux de réponse objective (TRO) confirmée† % (IC à 95 %)‡ |
41,8 (30,8 ; 53,4) |
|
Réponse complète, n (%) |
4 (5,1) |
|
Réponse partielle, n (%) |
29 (36,7) |
|
Durée de la réponse Médiane§, mois (IC à 95 %)¶ |
8,1 (5,9 ; NE) |
NE = non estimable.
* Inclut tous les patients ayant reçu au moins une dose d'Enhertu.
† Évaluée par revue centralisée indépendante.
‡ Calculé selon la méthode de Clopper-Pearson.
§ Selon une estimation de Kaplan-Meier.
¶ Calculé selon la méthode de Brookmeyer-Crowley.
* Inclut tous les patients ayant reçu au moins une dose d'Enhertu.
† Évaluée par revue centralisée indépendante.
‡ Calculé selon la méthode de Clopper-Pearson.
§ Selon une estimation de Kaplan-Meier.
¶ Calculé selon la méthode de Brookmeyer-Crowley.
DESTINY-Gastric01 (NCT03329690)
L'efficacité
et la sécurité d'Enhertu ont été évaluées dans l'étude DESTINY-Gastric01, une
étude de phase II multicentrique en ouvert, randomisée, menée au Japon et en
Corée du Sud. Dans cette étude supportive ont été inclus des patients adultes
présentant un adénocarcinome gastrique ou de la jonction œsogastrique (JOG) HER2-positif
localement avancé ou métastatique qui avait progressé sous au moins deux lignes
de traitement antérieures, comprenant le trastuzumab, une fluoropyrimidine et
un sel de platine. Les patients ont été
randomisés selon un rapport
2:1 pour recevoir Enhertu (N = 126) ou
la chimiothérapie choisie par le médecin : irinotécan (N = 55) ou paclitaxel (N
= 7). Les échantillons tumoraux devaient présenter une positivité HER2 confirmée
de manière centralisée, définie comme un score IHC 3+ ou un score IHC 2+/HIS+.
Les patients ayant des antécédents de PID/pneumopathie inflammatoire ayant nécessité une corticothérapie ou
présentant une PID/pneumopathie inflammatoire lors de la sélection, les patients
ayant des antécédents de cardiopathie cliniquement significative et les
patients présentant des métastases cérébrales évolutives étaient exclus de
l'étude. Le traitement était administré jusqu'à progression de la maladie,
décès, retrait du consentement ou survenue d'unetoxicité inacceptable. Le critère d'évaluation
principal de l'efficacité était le taux
de réponse objective (TRO) non confirmée, évaluée
par RCI selon les critères
RECIST v1.1. La survie globale
(SG) était le principal critère secondaire. La survie
sans progression (SSP), la durée de la réponse (DR) et le taux de réponse
objective confirmée étaient des critères d'évaluation secondaires
supplémentaires.
Les caractéristiques démographiques et cliniques des patients à l'inclusion étaient comparables entre les bras de traitement. Chez les 188 patients, l'âge médian était de 66 ans (plage : 28 à 82 ans), 76 % étaient des hommes, 100 % étaient asiatiques. Les patients avaient un indice de performance ECOG de 0 (49 %) ou 1 (51 %), 87 % présentaient un adénocarcinome de l'estomac et 13 % un adénocarcinome de la JOG, 76 % avaient un score IHC 3+ et 23 % un score IHC 2+/HIS+, 54 % présentaient des métastases hépatiques et 29 % des métastases pulmonaires ; la somme des diamètres des lésions cibles était < 5 cm chez 47 % des patients, ≥ 5 et < 10 cm chez 30 % et ≥ 10 cm chez 17 % ; 55 % des patients avaient reçu préalablement deux lignes de traitement et 45 % avaient reçu trois lignes de traitement ou plus pour la maladie localement avancée ou métastatique.
Les résultats d'efficacité (date de gel des données : 3 juin 2020) pour Enhertu (n = 126) versus chimiothérapie choisie par le médecin (n = 62) étaient un taux de réponse globale confirmée de 39,7 % (IC à 95 % : 31,1 ; 48,8) versus 11,3 % (IC à 95 % : 4,7 ; 21,9). Le taux de réponse complète était de 7,9 % versus 0 % et le taux de réponse partielle de 31,7 % versus 11,3 %. Les autres résultats d'efficacité pour Enhertu versus chimiothérapie choisie par le médecin étaient une DR médiane de 12,5 mois (IC à 95 % : 5,6 ; NE) versus 3,9 mois (IC à 95 % : 3,0 ; 4,9). La SSP médiane était de 5,6 mois (IC à 95 % : 4,3 ; 6,9) versus 3,5 mois (IC à 95 % : 2,0 ; 4,3 ; hazard ratio = 0,47 [IC à 95 % : 0,31 ; 0,71]). Une analyse de la SG, prédéfinie au moment où 133 décès auraient été observés, a montré un bénéfice en termes de survie dans le groupe traité par Enhertu par rapport au groupe recevant la chimiothérapie choisie par le médecin (hazard ratio = 0,60). La SG médiane était de 12,5 mois (IC à 95 % : 10,3 ; 15,2) dans le groupe traité par Enhertu et de 8,9 mois (IC à 95 % : 6,4 ; 10,4) dans le groupe recevant la chimiothérapie choisie par le médecin.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées dans tous les sous-groupes de la population pédiatrique dans l'indication de cancer du sein, de CBNPC et de cancer de l'estomac (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Une autorisation de mise sur le marché «
conditionnelle » a été délivrée
pour ce médicament. Cela
signifie que des preuves supplémentaires concernant ce médicament sont
attendues.
L'Agence européenne des médicaments réévaluera toute nouvelle
information sur ce médicament au moins chaque année et, si nécessaire,
ce RCP sera mis à
jour.
Absorption
Le trastuzumab déruxtécan est administré par voie intraveineuse. Il n'a pas été mené d'études avec d'autres voies d'administration.
Distribution
Selon une analyse pharmacocinétique de population, les volumes de distribution centraux (Vc) estimés du trastuzumab déruxtécan et de l'inhibiteur de la topoisomérase I, le DXd, sont respectivement de 2,68 litres et 28,0 litres.
In vitro, le taux moyen de liaison du DXd aux protéines plasmatiques humaines est d'environ 97 %.
In vitro, le rapport des concentrations sang/plasma du DXd est d'environ 0,6.
Biotransformation
Le trastuzumab déruxtécan est clivé dans la cellule par des enzymes lysosomales pour libérer le DXd.
Il est attendu que l'anticorps monoclonal IgG1 anti-HER2 humanisé soit dégradé en petits peptides et acides aminés par des voies cataboliques de la même façon que les IgG endogènes.
Des études de métabolisme in vitro sur microsomes hépatiques humains indiquent que le DXd est métabolisé principalement par le CYP3A4 par des voies oxydatives.
Élimination
Après administration par voie intraveineuse de trastuzumab déruxtécan chez des patients présentant un cancer du sein HER2-positif ou HER2-faible métastatique ou un CBNPC avec mutation d'HER2, la clairance du trastuzumab déruxtécan calculée dans une analyse pharmacocinétique de population est de 0,4 L/jour et celle du DXd est de 18,4 L/heure. Chez les patients présentant un adénocarcinome de l'estomac ou de la JOG localement avancé ou métastatique, la clairance du trastuzumab déruxtécan était plus élevée de 20 % que chez les patients présentant un cancer du sein HER2-positif métastatique. Lors du cycle 3, la demi-vie d'élimination apparente (t1/2) du trastuzumab déruxtécan et du DXd libéré est d'environ 7 jours. Une accumulation modérée du trastuzumab déruxtécan (environ 35 % lors du cycle 3 par rapport au cycle 1) a été observée.
Après administration de DXd par voie intraveineuse chez le rat, le produit était éliminé principalement dans les fèces par voie hépatobiliaire. Le DXd était le composant le plus abondant dans les urines, les fèces et la bile. Après administration unique de trastuzumab déruxtécan par voie intraveineuse (6,4 mg/kg) chez le singe, le DXd sous forme inchangée libéré était le composant le plus abondant dans les urines et les fèces. L'élimination du DXd n'a pas été étudiée chez l'homme.
Interactions in vitro
Effets d'Enhertu sur la
pharmacocinétique d'autres médicaments
Les
études in vitro indiquent que le DXd n'est pas
un inhibiteur des principales enzymes du CYP450 incluant les CYP1A2, 2B6, 2C8,
2C9, 2C19, 2D6 et 3A. Les études in vitro indiquent que le DXd n'est pas un inhibiteur des transporteurs OAT1, OAT3,
OCT1, OCT2, OATP1B1, OATP1B3, MATE1, MATE2-K, P-gp, BCRP ou BSEP.
Effets d'autres médicaments sur la pharmacocinétique d'Enhertu
In vitro, le DXd est un substrat
de la P-gp, d'OATP1B1, d'OATP1B3, de MATE2-K, de MRP1 et de la BCRP. Il n'est
pas attendu d'interactions cliniquement significatives avec les médicaments qui
sont des substrats des transporteurs MATE2-K, MRP1, P-gp, OATP1B ou BCRP (voir
rubrique Interactions avec d'autres médicaments et autres formes
d'interactions).
Linéarité/non-linéarité
Après administration par voie intraveineuse, l'exposition au trastuzumab déruxtécan et au DXd libéré augmente de façon proportionnelle à la dose dans la plage de doses de 3,2 mg/kg à 8,0 mg/kg (environ 0,6 à 1,5 fois la dose recommandée), avec une variabilité interindividuelle faible à modérée. Selon l'analyse pharmacocinétique de population, la variabilité interindividuelle était de 24 % et 28 % respectivement pour la clairance du trastuzumab déruxtécan et du DXd et de 16 % et 55 % respectivement pour le volume de distribution central. La variabilité intra-individuelle des valeurs de l'ASC (aire sous la courbe de la concentration sérique en fonction du temps) du trastuzumab déruxtécan et du DXd était d'environ 8 % et 14 % respectivement.
Populations particulières
Selon l'analyse pharmacocinétique de population, l'âge (20 à 96 ans), le groupe ethnique, l'ethnicité, le sexe et le poids corporel n'ont pas d'effet cliniquement significatif sur l'exposition au trastuzumab déruxtécan ou au DXd libéré.
Sujets âgés
L'analyse
pharmacocinétique de population a montré que l'âge (plage : 20 à 96 ans) n'a
pas d'effet sur la pharmacocinétique du trastuzumab déruxtécan.
Insuffisance rénale
Il
n'a pas été mené d'étude spécifique chez des patients présentant une
insuffisance rénale. Selon l'analyse pharmacocinétique de population ayant
inclus des patients présentant une insuffisance rénale légère (clairance de la
créatinine [ClCr] ≥ 60 et < 90 mL/min) ou modérée (ClCr ≥
30 et < 60 mL/min) (estimée selon la formule de
Cockcroft-Gault), la pharmacocinétique du DXd libéré
n'est pas modifiée en cas d'insuffisance rénale légère ou modérée par rapport
aux sujets ayant une fonction rénale normale (ClCr ≥
90 mL/min).
Insuffisance hépatique
Il
n'a pas été mené d'étude spécifique chez des patients présentant une
insuffisance hépatique. Selon l'analyse pharmacocinétique de population,
l'impact des modifications de la pharmacocinétique du trastuzumab
déruxtécan chez les patients ayant un taux de
bilirubine totale ≤ 1,5 × LSN, quel que soit le taux d'ASAT, n'est pas
cliniquement significatif. Les données chez les patients ayant un taux
de
bilirubine totale > 1,5 à 3 × LSN, quel que soit le taux d'ASAT,
sont limitées et ne permettent pas de tirer des conclusions et il
n'existe pas de
données chez les patients ayant un taux de bilirubine totale > 3 ×
LSN, quel
que soit le taux d'ASAT (voir rubriques Posologie et mode d'administration
et Mises en garde spéciales et précautions d'emploi).
Population pédiatrique
Il
n'a pas été mené d'études pour évaluer la pharmacocinétique du trastuzumab déruxtécan chez les
enfants ou les adolescents.
Enhertu peut avoir une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Les patients doivent être informés qu'ils doivent faire preuve de prudence lors de la conduite de véhicules ou de l'utilisation de machines en cas de fatigue, de céphalées ou de sensations vertigineuses pendant le traitement par Enhertu (voir rubrique Effets indésirables).
Chez l'animal, des effets toxiques ont été observés dans les organes lymphatiques et hématopoïétiques, l'intestin, les reins, les poumons, les testicules et la peau après l'administration de trastuzumab déruxtécan à des doses entraînant des niveaux d'exposition à l'inhibiteur de la topoisomérase I (DXd) inférieurs à l'exposition plasmatique chez l'homme. Chez ces animaux, l'exposition à l'anticorps conjugué était similaire ou supérieure à l'exposition plasmatique chez l'homme.
Le DXD a été clastogène dans un essai des micronoyaux sur cellules de moelle osseuse in vivo chez le rat et dans un essai d'aberrations chromosomiques sur cellules de poumon de hamster chinois in vitro et n'a pas été mutagène dans un essai de mutation réverse sur bactéries in vitro.
Il n'a pas été réalisé d'études de cancérogenèse avec le trastuzumab déruxtécan.
Il n'a pas été réalisé d'études spécifiques de la fertilité avec le trastuzumab déruxtécan. Selon les résultats des études de toxicologie générale effectuées chez l'animal, le trastuzumab déruxtécan peut altérer les fonctions de reproduction et la fertilité chez les mâles.
Il n'a pas été mené d'études de toxicité sur la reproduction ou le développement chez l'animal avec le trastuzumab déruxtécan. Selon les résultats des études de toxicologie générale effectuées chez l'animal, le trastuzumab déruxtécan et le DXd étaient toxiques pour les cellules à division rapide (organes lymphatiques/hématopoïétiques, intestin ou testicules) et le DXd était génotoxique, ce qui semble indiquer un potentiel d'embryotoxicité et de tératogénicité.
Afin d'éviter des erreurs médicamenteuses, il est important de vérifier l'étiquette du flacon pour s'assurer que le médicament préparé et administré est Enhertu (trastuzumab déruxtécan) et non le trastuzumab ou le trastuzumab emtansine.
Des procédures appropriées pour la préparation des médicaments cytotoxiques doivent être utilisées. Une technique aseptique appropriée doit être utilisée pour les procédures de reconstitution et de dilution ci-dessous.
Reconstitution
- La reconstitution doit être effectuée immédiatement avant la dilution.
- Plusieurs flacons peuvent être nécessaires pour obtenir la pleine dose. Calculer la dose (en mg), le volume total de solution d'Enhertu reconstituée nécessaire et le nombre de flacons d'Enhertu nécessaires (voir rubrique Posologie et mode d'administration).
- Reconstituer chaque flacon de 100 mg en utilisant une seringue stérile pour injecter lentement 5 mL d'eau pour préparations injectables dans chaque flacon afin d'obtenir une concentration finale de 20 mg/mL.
- Faire tourner doucement le flacon jusqu'à dissolution complète. Ne pas agiter.
- D'un point de vue microbiologique, le produit doit être utilisé immédiatement. Dans le cas contraire, la stabilité physico-chimique après reconstitution a été démontrée pendant une durée allant jusqu'à 48 heures à une température comprise entre 2 °C et 8 °C. Conserver les flacons contenant la solution d'Enhertu reconstituée au réfrigérateur entre 2 °C et 8 °C à l'abri de la lumière. Ne pas congeler.
- Le médicament reconstitué ne contient pas de conservateur et est à usage unique.
Dilution
- Prélever la quantité calculée du ou des flacons à l'aide d'une seringue stérile. Examiner la solution reconstituée pour vérifier l'absence de particules et de coloration anormale. La solution doit être limpide et incolore à jaune clair. Ne pas utiliser si la solution contient des particules visibles ou si elle est trouble ou présente une coloration anormale.
- Diluer le volume calculé de solution d'Enhertu reconstituée dans une poche à perfusion contenant 100 mL de solution pour perfusion de glucose à 5 %. Ne pas utiliser de solution de chlorure de sodium (voir rubrique Incompatibilités). L'utilisation de poches à perfusion en polychlorure de vinyle ou en polyoléfine (copolymère d'éthylène et de polypropylène) est recommandée.
- Retourner doucement la poche à perfusion pour mélanger complètement la solution. Ne pas agiter.
- Couvrir la poche à perfusion pour la protéger de la lumière.
- Si le médicament n'est pas utilisé immédiatement, conserver à température ambiante (≤ 30 ºC) pendant 4 heures au maximum en incluant les durées de préparation et de perfusion ou au réfrigérateur entre 2 °C et 8 °C pendant 24 heures au maximum, à l'abri de la lumière. Ne pas congeler.
- Éliminer toute solution non utilisée restant dans le flacon.
Administration
- Si la solution pour perfusion préparée a été conservée au réfrigérateur (entre 2 °C et 8 °C), il est recommandé de la laisser s'équilibrer à température ambiante à l'abri de la lumière avant l'administration.
- Administrer Enhertu en perfusion intraveineuse stricte, avec un filtre en ligne en polyéthersulfone (PES) ou en polysulfone (PS) de 0,20 ou 0,22 micron.
- La dose initiale doit être administrée en perfusion intraveineuse de 90 minutes. Si la première perfusion a été bien tolérée, les doses suivantes d'Enhertu peuvent être administrées en perfusion de 30 minutes. Ne pas administrer en injection intraveineuse rapide ou en bolus (voir rubrique Posologie et mode d'administration).
- Couvrir la poche à perfusion pour la protéger de la lumière.
- Ne pas mélanger Enhertu avec d'autres médicaments ni administrer d'autres médicaments par la même ligne intraveineuse.
Élimination
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription réservée aux spécialistes en oncologie ou aux médecins compétents en cancérologie.
Réservé à l'usage HOSPITALIER.
Prescription réservée aux spécialistes en oncologie ou aux médecins compétents en cancérologie.
Réservé à l'usage HOSPITALIER.
Poudre pour solution à diluer pour perfusion.
Poudre lyophilisée de couleur blanche à blanc-jaunâtre.
Enhertu est présenté en flacon en verre borosilicate jaune de type I de 10 mL fermé par un bouchon en laminé de caoutchouc butyle et résine fluorée avec opercule serti jaune en polypropylène/aluminium. Chaque boîte contient un flacon.
Un flacon de poudre pour solution à diluer pour perfusion contient 100 mg de trastuzumab déruxtécan. Après reconstitution, un flacon de 5 mL de solution contient 20 mg/mL de trastuzumab déruxtécan (voir rubrique Précautions particulières d'élimination et de manipulation).
Le trastuzumab déruxtécan est un anticorps conjugué contenant un anticorps monoclonal (ACm) de type IgG1 anti-HER2 humanisé ayant la même séquence d'acides aminés que le trastuzumab, produit dans des cellules de mammifère (cellules d'ovaire de hamster chinois), lié de façon covalente au DXd, un dérivé de l'exatécan inhibiteur de la topoisomérase I, par un agent de liaison tétrapeptidique clivable. Environ 8 molécules de déruxtécan sont liées à chaque molécule d'anticorps.
Excipient à effet notoire :
Chaque flacon de 100 mg contient 1,5 mg de polysorbate 80 (E 433).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
L-histidine
Chlorhydrate de L-histidine monohydraté
Saccharose
Polysorbate 80 (E 433)